医薬品情報
| 総称名 | ビルベイ |
|---|---|
| 一般名 | オデビキシバット水和物 |
| 欧文一般名 | Odevixibat Hydrate |
| 製剤名 | オデビキシバット水和物顆粒 |
| 薬効分類名 | 回腸胆汁酸トランスポーター阻害剤 |
| 薬効分類番号 | 3919 |
| KEGG DRUG |
D13092
オデビキシバット水和物
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年4月 改訂(第3版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ビルベイ顆粒200μg | Bylvay Granules | IPSEN | 3919009D1020 | 29705.1円/個 | 劇薬, 処方箋医薬品注) |
| ビルベイ顆粒600μg | Bylvay Granules | IPSEN | 3919009D2027 | 89114.7円/個 | 劇薬, 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 妊婦又は妊娠している可能性のある女性[9.5参照]
4. 効能または効果
進行性家族性肝内胆汁うっ滞症に伴うそう痒
5. 効能または効果に関連する注意
ABCB11遺伝子変異を有する患者のうち、胆汁酸塩排出ポンプ蛋白質(BSEP)の機能を完全に喪失する変異を有する患者では、本剤の効果は期待できない。
6. 用法及び用量
通常、オデビキシバットとして40μg/kgを1日1回朝食時に経口投与する。なお、効果不十分な場合には、120μg/kgを1日1回に増量することができるが、1日最高用量として7200μgを超えないこと。
7. 用法及び用量に関連する注意
7.1 本剤によるそう痒の改善や血清中胆汁酸濃度の低下は緩徐に認められることがあるため、増量の判断は投与開始3ヵ月以降とし、忍容性に問題がない場合に行うこと。また、本剤を6ヵ月間投与しても効果が認められない場合には、投与継続の是非を検討すること。
7.2 体重別の1日投与量は下表を参考にすること。
40μg/kg/日の場合
| 体重(kg) | 1日投与量(μg) |
| 5.0以上7.5未満 | 200 |
| 7.5以上12.5未満 | 400 |
| 12.5以上17.5未満 | 600 |
| 17.5以上25.5未満 | 800 |
| 25.5以上35.5未満 | 1200 |
| 35.5以上45.5未満 | 1600 |
| 45.5以上55.5以下 | 2000 |
| 55.5超 | 2400 |
120μg/kg/日の場合
| 体重(kg) | 1日投与量(μg) |
| 5.0以上7.5未満 | 600 |
| 7.5以上12.5未満 | 1200 |
| 12.5以上17.5未満 | 1800 |
| 17.5以上25.5未満 | 2400 |
| 25.5以上35.5未満 | 3600 |
| 35.5以上45.5未満 | 4800 |
| 45.5以上55.5以下 | 6000 |
| 55.5超 | 7200 |
7.3 カプセルは容器であることから、カプセルごと投与せず、容器内の顆粒剤のみを全量投与すること。[14.1参照]
7.4 本剤は、1日の最初の食事の際に飲食物とともに投与すること。[14.1参照]
8. 重要な基本的注意
8.1 肝機能検査値の上昇がみられることがあるので、本剤の投与開始前及び投与期間中は定期的に肝機能検査を行い、患者の状態を十分に観察すること。
8.2 下痢があらわれることがあり、脱水症状を引き起こす可能性がある。本剤投与中に腹痛や下痢が持続し、他の原因が認められない場合は、減量又は投与の中断若しくは中止を考慮すること。下痢による脱水に注意し、異常が認められた場合には速やかに適切な処置を行うこと。
8.3 脂溶性ビタミンの減少がみられることがあるので、本剤の投与開始前及び投与期間中は定期的に血中脂溶性ビタミン(ビタミンA、D、E、K)濃度及びプロトロンビン時間国際標準比(PT-INR)を測定し、患者の状態を十分に観察し、必要に応じて脂溶性ビタミンの補充を考慮すること。
9. 特定の背景を有する患者に関する注意
9.3 肝機能障害患者
9.3.1 重度の肝機能障害患者(Child Pugh 分類C)
患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。血中濃度が上昇するおそれがある。重度の肝機能障害患者を対象とした臨床試験は実施していない。
9.4 生殖能を有する者
妊娠する可能性のある女性には、本剤投与中及び最終投与後5日間において避妊する必要性及び適切な避妊法について説明すること。[9.5参照]
9.5 妊婦
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。ヒトにおける乳汁中への移行に関するデータはないが、動物実験(ラット)で、母動物へ投与後授乳された乳児への曝露が認められている1)。
9.7 小児等
体重5kg未満の小児等を対象とした有効性及び安全性を指標とした臨床試験は実施していない。
11. 副作用
11.2 その他の副作用
| 10%以上 | 1%以上10%未満 | |
| 肝胆道系障害 | 血中ビリルビン増加、ALT増加 | 肝腫大、AST増加 |
| 胃腸障害 | 下痢、嘔吐、腹痛 | |
| 代謝および栄養障害 | ビタミンD欠乏 | ビタミンE欠乏 |
14. 適用上の注意
14.1 薬剤交付時の注意
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
外国人健康成人にオデビキシバット0.1、0.3、1、3及び10mgを単回経口投与したとき、3mgを投与した6例中2例、10mgを投与した6例中5例で定量可能な血漿中薬物濃度[定量下限=0.05ng/mL]が認められた。10mgを投与したときのCmax及びAUC0-tの幾何平均値[幾何変動係数(CV%)]はそれぞれ0.16ng/mL(85.6%)及び0.58ng・h/mL(39.0%)であり、Tmaxの中央値は4.04時間であった2)。
日本人健康成人にオデビキシバット3mgを単回経口投与したとき、投与後1.5〜8時間で血漿中薬物濃度は6例中5例に検出された。Cmax及びAUC0-lastの平均値(標準偏差)はそれぞれ0.06385(0.03223)ng/mL及び0.29(0.10)h・ng/mLであり、Tmaxの中央値は6.000時間であった3)。
日本人健康成人にオデビキシバット3mgを単回経口投与したとき、投与後1.5〜8時間で血漿中薬物濃度は6例中5例に検出された。Cmax及びAUC0-lastの平均値(標準偏差)はそれぞれ0.06385(0.03223)ng/mL及び0.29(0.10)h・ng/mLであり、Tmaxの中央値は6.000時間であった3)。
16.1.2 反復投与
16.2 吸収
16.2.1 投与方法の影響
高脂肪食(800〜1,000キロカロリーで食事の総カロリー量の約50%が脂肪)とカプセル剤の同時投与は、カプセル剤の空腹時の投与と比較してCmaxが約72%、AUC0-24が約62%低下した。オデビキシバットの顆粒剤をアップルソースに振りかけた場合、カプセル剤の空腹時投与と比較して、Cmaxが約39%、AUC0-24が約36%低下した4)(外国人データ)。
16.2.2 バイオアベイラビリティ
オデビキシバットは経口投与後ほとんど吸収されない。ヒトにおける絶対的バイオアベイラビリティのデータはない。最高血漿濃度(Cmax)は1〜5時間以内に達する。小児患者(年齢0.8〜16.0歳、体重5.6〜55.2kg)のトラフ値は、40μg/kg/日及び120μg/kg/日の患者の83%及び80%の検体で検出限界以下であった。
母集団薬物動態解析から推定された小児進行性家族性肝内胆汁うっ滞症患者における40μg/kg/日及び120μg/kg/日の投与量のCmaxの値はそれぞれ0.211ng/mL及び0.623ng/mLであり、AUC値はそれぞれ2.26ng・h/mL及び5.99ng・h/mLであった5)(外国人データ)。
母集団薬物動態解析から推定された小児進行性家族性肝内胆汁うっ滞症患者における40μg/kg/日及び120μg/kg/日の投与量のCmaxの値はそれぞれ0.211ng/mL及び0.623ng/mLであり、AUC値はそれぞれ2.26ng・h/mL及び5.99ng・h/mLであった5)(外国人データ)。
16.3 分布
16.4 代謝
ヒト肝細胞における代謝割合は、8.2〜22.6%であった6)。
16.5 排泄
16.7 薬物相互作用
16.7.1 エチニルエストラジオール、レボノルゲストレル
健康女性被験者(25例)にオデビキシバット3mgと経口避妊薬(エチニルエストラジオール(EE)0.03mg/レボノルゲストレル(LVN)0.15mg)を単回併用投与したとき、オデビキシバット併用時の非併用時に対するEEのAUC0-infは83%、LVNのAUC0-infは88%であった8)(外国人データ)。
16.7.2 ミダゾラム
健康被験者(20例)を対象にオデビキシバット7.2mgにミダゾラム2mgを単回併用投与したとき、オデビキシバット併用時の非併用時に対するミダゾラムのAUC0-infは72%、1-OHミダゾラムのAUC0-infは86%であった9)(外国人データ)。
16.7.3 イトラコナゾール
健康被験者(21例)にイトラコナゾール200mgとオデビキシバット7.2mgを単回併用投与したとき、イトラコナゾール併用時の非併用時に対するオデビキシバットのAUC0-infは151%であった9)(外国人データ)。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内第III相試験
非盲検第III相試験において、日本人進行性家族性肝内胆汁うっ滞症患者(体重5kg超)にオデビキシバット40μg/kgを1日1回12週間、12週以降は安全性に問題がない場合120μg/kgを1日1回、24週以降は40μg/kg又は120μg/kg(最大7200μg)を1日1回経口投与した。
なお、同意取得時に生後6ヵ月以上18歳未満で、PFIC-1又はPFIC-2と診断されている患者をコホート1(2例)、コホート1に該当しない生後3ヵ月以上のPFIC患者又はコホート1の組み入れ期間終了後の患者をコホート2(1例)とした。
主要評価項目は、ベースラインから24週時までに空腹時血清中胆汁酸濃度が70%以上低下又は24週時に70μmol/L以下に到達した患者の割合、及び24週間における患者レベルでの痒み評価が改善した割合とした。
ベースラインから24週時までに空腹時血清中胆汁酸濃度が70%以上低下又は24週時に70μmol/L以下に到達した患者の割合は、コホート1では50.0%(1/2例)、コホート2では0.0%(0/1例)であり、24週間における患者レベルでの痒み評価が改善した割合は、コホート1の2例は77.7%及び0.3%、コホート2の1例は77.4%であった。
コホート1及びコホート2で副作用は認められなかった(48週)10)。
なお、同意取得時に生後6ヵ月以上18歳未満で、PFIC-1又はPFIC-2と診断されている患者をコホート1(2例)、コホート1に該当しない生後3ヵ月以上のPFIC患者又はコホート1の組み入れ期間終了後の患者をコホート2(1例)とした。
主要評価項目は、ベースラインから24週時までに空腹時血清中胆汁酸濃度が70%以上低下又は24週時に70μmol/L以下に到達した患者の割合、及び24週間における患者レベルでの痒み評価が改善した割合とした。
ベースラインから24週時までに空腹時血清中胆汁酸濃度が70%以上低下又は24週時に70μmol/L以下に到達した患者の割合は、コホート1では50.0%(1/2例)、コホート2では0.0%(0/1例)であり、24週間における患者レベルでの痒み評価が改善した割合は、コホート1の2例は77.7%及び0.3%、コホート2の1例は77.4%であった。
コホート1及びコホート2で副作用は認められなかった(48週)10)。
17.1.2 海外第III相試験
無作為化二重盲検プラセボ対照第III相試験において、6ヵ月以上18歳以下かつ体重5kg超の進行性家族性肝内胆汁うっ滞症(PFIC-1又はPFIC-2)患者(62例)を対象に、オデビキシバット40μg/kg、120μg/kg(最大7200μg)又はプラセボを1日1回24週間経口投与した。
主要評価項目は、24週時までに空腹時血清中胆汁酸濃度がベースラインから70%以上低下又は24週時に70μmol/L以下に達した患者の割合とした。24週間の投与期間にわたる観察者報告アウトカム(ObsRO)尺度に基づく患者レベルでの痒み評価が改善した割合を副次的評価項目とし、結果は下表のとおりであった。
主要評価項目は、24週時までに空腹時血清中胆汁酸濃度がベースラインから70%以上低下又は24週時に70μmol/L以下に達した患者の割合とした。24週間の投与期間にわたる観察者報告アウトカム(ObsRO)尺度に基づく患者レベルでの痒み評価が改善した割合を副次的評価項目とし、結果は下表のとおりであった。
| 有効性評価項目 | プラセボ群 (20例) | オデビキシバット群 | ||
| 40μg/kg/日 (23例) | 120μg/kg/日 (19例) | 合計 (42例) | ||
| 24週時に血清中胆汁酸濃度が低下した患者の割合 | ||||
| 例数(%) (95%信頼区間) | 0(0.0) (0.0,16.8) | 10(43.5) (23.2,65.5) | 4(21.1) (6.1,45.6) | 14(33.3) (19.6,49.6) |
| プラセボとの差 (95%信頼区間)a) | − | 44.1 (23.6,64.6) | 21.6 (−0.5,43.8) | 30.7 (12.6,48.8) |
| 片側p値b) | − | 0.0003 | 0.0174 | 0.0015 |
| 24週間における患者レベルでの痒み評価が改善した割合 | ||||
| 改善割合% 平均値±SD | 28.7±5.2 | 58.3±6.2 | 47.7±8.1 | 53.5±5.0 |
| プラセボとの差 (95%信頼区間)c) | − | 28.2 (9.8,46.6) | 21.7 (1.9,41.5) | 25.0 (8.5,41.5) |
副作用発現率は本剤40μg/kg群で30.4%(7/23例)、120μg/kg群で36.8%(7/19例)であった。主な副作用は40μg/kg群で血中ビリルビン増加、アラニンアミノトランスフェラーゼ増加、アスパラギン酸アミノトランスフェラーゼ増加、及び下痢が各8.7%(各2/23例)、120μg/kg群で血中ビリルビン増加、アラニンアミノトランスフェラーゼ増加、及び下痢が各10.5%(各2/19例)であった11)。
17.1.3 海外第III相継続投与試験
非盲検第III相継続投与試験において、海外第III相試験から移行した進行性家族性肝内胆汁うっ滞症患者(コホート1:56例(海外第III相試験でオデビキシバット群37例、プラセボ群19例))、及び年齢不問かつ体重5kg以上で、血清中胆汁酸濃度の上昇及び胆汁うっ滞性そう痒を伴う進行性家族性肝内胆汁うっ滞症(病型不問)患者(コホート2:60例)を対象に、オデビキシバット40μg/kg又は120μg/kgを1日1回72週間経口投与した。
主要評価項目は、72週時の空腹時血清中胆汁酸濃度のベースラインからの変化とした。72週時までの投与期間における観察者報告アウトカム(ObsRO)尺度に基づく患者レベルでの痒み評価が改善した割合を副次的評価項目とし、結果は下表のとおりであった。
主要評価項目は、72週時の空腹時血清中胆汁酸濃度のベースラインからの変化とした。72週時までの投与期間における観察者報告アウトカム(ObsRO)尺度に基づく患者レベルでの痒み評価が改善した割合を副次的評価項目とし、結果は下表のとおりであった。
| 有効性評価項目 | コホート1 | コホート2 60例 | |
| プラセボ/オデビキシバット 19例 | オデビキシバット/オデビキシバット 37例 | ||
| 72週時の空腹時血清中胆汁酸濃度(μmol/L)のベースラインa)からの変化 | |||
| 例数 | 15 | 28 | 43 |
| 平均変化量 (SD) | −104.00 (167.32) | −139.84 (172.07) | −57.97 (137.99) |
| 変化率%の中央値 | −18.04 | −58.47 | −24.83 |
| 72週間における患者レベルでの痒み評価が改善した割合 | |||
| 例数 | 12 | 26 | 31 |
| 平均値%±SD | 55.2±38.7 | 38.6±34.9 | 77.3±28.1 |
副作用発現率は38.8%(45/116例)であった。主な副作用は下痢12.1%(14/116例)、血中ビリルビン増加10.3%(12/116例)、及びアラニンアミノトランスフェラーゼ増加6.0%(7/116例)であった12)。
18. 薬効薬理
18.1 作用機序
オデビキシバットは、回腸胆汁酸トランスポーター(IBAT)の可逆的かつ強力な選択的阻害剤である。遠位回腸に局所的に作用して、胆汁酸(主に胆汁酸塩の形態)の再取り込みを減少させ、結腸を通過する胆汁酸のクリアランスを増加させ、血清中の胆汁酸濃度を低下させる。
19. 有効成分に関する理化学的知見
19.1. オデビキシバット水和物
| 一般的名称 | オデビキシバット水和物 |
|---|---|
| 一般的名称(欧名) | Odevixibat Hydrate |
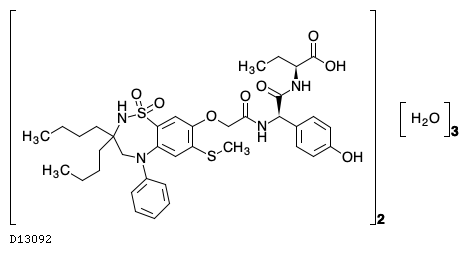
| 化学名 | (2S)-2-[(2R)-2-(2-{[3,3-Dibutyl-7-(methylsulfanyl)-1,1-dioxo-5-phenyl-2,3,4,5-tetrahydro-1H-1λ6,2,5-benzothiadiazepin-8-yl]oxy}acetamido)-2-(4-hydroxyphenyl)acetamido]butanoic acid sesquihydrate |
| 分子式 | C37H48N4O8S2・11/2H2O |
| 分子量 | 767.95 |
| 物理化学的性状 | オデビキシバット水和物は白色〜オフホワイトの粉末である。水溶液中の溶解度はpHに依存し、pHの上昇とともに増加する。 |
| KEGG DRUG |
|
20. 取扱い上の注意
光を避けるため、ボトル開封後も元のボトルのまま保管すること。
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
<ビルベイ顆粒200μg>
30カプセル型容器[ボトル]
<ビルベイ顆粒600μg>
30カプセル型容器[ボトル]
23. 主要文献
- 社内資料:生殖発生毒性試験(2025年9月19日承認、CTD2.6.6.6)
- 社内資料:単回及び反復漸増試験(A4250-001試験)(2025年9月19日承認、CTD2.7.2.2.2.1)
- 社内資料:日本人健康成人被験者での反復投与試験(A4250-J001試験)(2025年9月19日承認、CTD2.7.2.2.2.2)
- 社内資料:食事の影響及び振りかけ試験(A4250-004試験)(2025年9月19日承認、CTD2.7.2.2.2.4)
- 社内資料:母集団PK解析(2025年9月19日承認、CTD2.7.2.3.1)
- 社内資料:ヒト肝細胞による代謝試験(2025年9月19日承認CTD2.7.2.2.1.4)
- 社内資料:14C-ADME/マスバランス試験(A4250-007試験)(2025年9月19日承認、CTD2.7.2.2.2.7)
- 社内資料:薬物間相互作用試験(A4250-022試験)(2025年9月19日承認、CTD2.7.2.2.2.9)
- 社内資料:薬物間相互作用試験(A4250-013試験)(2025年9月19日承認、CTD2.7.2.2.2.8)
- 社内資料:オデビキシバット(A4250)の進行性家族性肝内胆汁うっ滞症患者における安全性及び有効性を評価する非盲検第III相試験(A4250-J005試験)(2025年9月19日承認、CTD2.7.6.9)
- 社内資料:進行性家族性肝内胆汁うっ滞症1型及び2型(PFIC-1及びPFIC-2)の小児患者を対象とする第III相試験(A4250-005試験)(2025年9月19日承認、CTD2.7.6.8)
- 社内資料:進行性家族性肝内胆汁うっ滞症(PFIC)の小児患者を対象とした第III相継続投与試験(A4250-008試験)(2025年9月19日承認、CTD2.7.6.10)
24. 文献請求先及び問い合わせ先
文献請求先
IPSEN株式会社
製品情報担当
〒100-6162
東京都千代田区永田町二丁目11番1号 山王パークタワー3階
電話:03-6205-3483
製品情報問い合わせ先
IPSEN株式会社
製品情報担当
〒100-6162
東京都千代田区永田町二丁目11番1号 山王パークタワー3階
電話:03-6205-3483
26. 製造販売業者等
26.1 製造販売元
IPSEN株式会社
東京都千代田区永田町二丁目11番1号 山王パークタワー3階