医薬品情報
| 総称名 | ヘルネクシオス |
|---|---|
| 一般名 | ゾンゲルチニブ |
| 欧文一般名 | Zongertinib |
| 製剤名 | ゾンゲルチニブ |
| 薬効分類名 | 抗悪性腫瘍剤 HER2注1)阻害剤 注1)HER2:Human Epidermal Growth Factor Receptor Type 2(ヒト上皮増殖因子受容体2型、別称:c-erbB-2) |
| 薬効分類番号 | 4291 |
| KEGG DRUG |
D12879
ゾンゲルチニブ
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年11月 改訂(第2版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ヘルネクシオス錠60mg | HERNEXEOS Tablets 60mg | 日本ベーリンガーインゲルハイム | 4291096F1020 | 13881.9円/錠 | 劇薬, 処方箋医薬品注2) |
1. 警告
本剤は、緊急時に十分対応できる医療施設において、がん化学療法に十分な知識・経験を持つ医師のもとで、本剤の投与が適切と判断される症例についてのみ投与すること。また、治療開始に先立ち、患者又はその家族に有効性及び危険性を十分説明し、同意を得てから投与すること。
2. 禁忌
次の患者には投与しないこと
本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
がん化学療法後に増悪したHER2(ERBB2)遺伝子変異陽性の切除不能な進行・再発の非小細胞肺癌
5. 効能または効果に関連する注意
5.1 十分な経験を有する病理医又は検査施設における検査により、HER2(ERBB2)遺伝子変異が確認された患者に投与すること。検査にあたっては、承認された体外診断用医薬品又は医療機器を用いること。なお、承認された体外診断用医薬品又は医療機器に関する情報については、以下のウェブサイトから入手可能である:
https://www.pmda.go.jp/review-services/drug-reviews/review-information/cd/0001.html
5.2 本剤の一次治療における有効性及び安全性は確立していない。
5.3 本剤の術前・術後補助療法における有効性及び安全性は確立していない。
5.4 「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、本剤以外の治療の実施についても慎重に検討し、適応患者の選択を行うこと。[17.1.1参照]
6. 用法及び用量
通常、成人には、ゾンゲルチニブとして1日1回120mgを経口投与する。なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
7.1 他の抗悪性腫瘍剤との併用について、有効性及び安全性は確立していない。
7.2 本剤投与により副作用が発現した場合には、以下の基準を参考に本剤を休薬、減量又は中止すること。1日1回60mgに減量しても忍容性が認められない場合は、本剤の投与を中止すること。
| 副作用 | 重症度注) | 処置 |
| 肝機能障害 | Grade 3又は4のALT又はAST増加 | Grade 1以下又はベースラインに回復するまで休薬し、回復後は60mgで再開できる。 |
| Grade 3の総ビリルビン増加 | Grade 1以下又はベースラインに回復するまで休薬し、回復後は60mgで再開できる。 | |
| Grade 4の総ビリルビン増加 | 投与を中止する。 | |
| ALT又はASTが基準値上限の3倍以上かつ総ビリルビンが基準値上限の2倍以上 | 投与を中止する。 | |
| 下痢 | Grade 2かつ止瀉薬の投与を行っても症状が2日以上継続する場合 | Grade 1以下に回復するまで休薬し、回復後は60mgで再開できる。 |
| Grade 3又は4 | Grade 1以下に回復するまで休薬する。 14日以内に回復した場合は60mgで再開できる。支持療法を行っても14日以内に回復しない場合は、投与を中止する。 | |
| 発熱性好中球減少症 | 全Grade | Grade 1以下又はベースラインに回復するまで休薬し、回復後は60mgで再開できる。ただし、必要に応じて投与を中止することも考慮する。 |
| 駆出率減少 | Grade 2 | Grade 1以下に回復するまで休薬し、回復後は120mgで再開できる。ただし、休薬後4週間以内に正常範囲又はベースラインから5ポイント以内に回復しなかった場合、再開後にベースラインから10ポイント以上低下した場合は60mgに減量する。 |
| Grade 3又は4 | Grade 1以下に回復するまで休薬し、回復後は60mgで再開できる。ただし、休薬後4週間以内に正常範囲又はベースラインから5ポイント以内に回復しなかった場合、再開後にベースラインから10ポイント以上低下した場合は投与を中止する。 | |
| 間質性肺疾患 | Grade 2 | Grade 1以下に回復するまで休薬する。14日以内に回復した場合は60mgで再開できる。支持療法を行っても14日以内にGrade 1以下に回復しない場合は投与を中止する。 |
| Grade 3又は4 | 投与を中止する。 | |
| その他の副作用 | Grade 3又は4 | Grade 1以下又はベースラインに回復するまで休薬し、回復後は60mgで再開できる。ただし、必要に応じて投与を中止することも考慮する。 |
8. 重要な基本的注意
8.1 肝機能障害があらわれることがあるので、本剤投与開始前及び投与中は定期的に肝機能検査を行い、患者の状態を十分に観察すること。[11.1.2参照]
8.2 血球減少があらわれることがあるので、本剤投与開始前及び投与中は定期的に血液検査を行い、患者の状態を十分に観察すること。[11.1.3参照]
8.3 間質性肺疾患があらわれることがあるので、初期症状(呼吸困難、咳嗽、発熱等)の確認及び胸部画像検査の実施等、患者の状態を十分に観察すること。また、患者に対して、初期症状があらわれた場合には、速やかに医療機関を受診するよう説明すること。[11.1.4参照]
8.4 左室駆出率(LVEF)低下があらわれることがあるので、本剤投与開始前及び本剤投与中は適宜心機能検査(心エコー等)を行い、患者の状態(LVEFの変動を含む)を十分に観察すること。[9.1.1参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 左室駆出率(LVEF)が低下している患者
LVEF低下を悪化させるおそれがある。[8.4参照]
9.3 肝機能障害患者
9.3.1 中等度以上の肝機能障害を有する患者
本剤は主に肝臓で代謝されるため、血中濃度が上昇する可能性がある。なお、中等度以上の肝機能障害のある患者注)を対象とした臨床試験は実施していない。
注)NCI-ODWG(National Cancer Institute-Organ Dysfunction Working Group)基準による分類
9.4 生殖能を有する者
9.4.1 妊娠する可能性のある女性には、本剤投与中及び最終投与後10日間において避妊する必要性及び適切な避妊法について説明すること。[9.5参照]
9.4.2 生殖可能な女性に投与する場合には、受胎能の低下があらわれる可能性があることを考慮すること。動物実験(ラット)において、子宮の萎縮、並びに子宮頸部及び膣の過形成/過角化が報告されている。
9.5 妊婦
9.6 授乳婦
授乳しないことが望ましい。本剤が乳汁に移行する可能性があり、乳児が乳汁を介して摂取した場合、乳児に重篤な副作用が発現するおそれがある。[9.5参照]
9.7 小児等
小児等を対象とした本剤の有効性及び安全性は確立していない。
10. 相互作用
相互作用序文
本剤は、主にCYP3Aによって代謝され、P-gp及びBCRPの阻害作用を示す。
薬物代謝酵素用語
CYP3A
薬物代謝酵素用語
P-gp
薬物代謝酵素用語
BCRP
10.2 併用注意
| 強いCYP3A誘導剤 カルバマゼピン、リファンピシン、フェニトイン等 [16.7.1参照] | 本剤の有効性が減弱するおそれがあるため、これらの薬剤との併用は可能な限り避け、CYP3A誘導作用のない又は弱い薬剤への代替を考慮すること。 | これらの薬剤がCYP3Aを誘導することにより、本剤の血中濃度が低下する可能性がある。 |
| 治療域の狭いP-gpの基質となる薬剤 シクロスポリン、エベロリムス、シロリムス等 [16.7.1参照] | これらの薬剤の副作用が増強されるおそれがあるため、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | 本剤がP-gpを阻害することにより、これらの薬剤の血中濃度が上昇する可能性がある。 |
| BCRPの基質となる薬剤 ロスバスタチン、メトトレキサート、サラゾスルファピリジン等 [16.7.1参照] | これらの薬剤の副作用が増強されるおそれがあるため、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | 本剤がBCRPを阻害することにより、これらの薬剤の血中濃度が上昇する可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 重度の下痢(3.0%)
11.1.2 肝機能障害(35.2%)[8.1参照]
11.1.3 血球減少
発熱性好中球減少症(頻度不明)、好中球減少症(0.8%)、貧血(17.8%)、血小板減少症(0.8%)等があらわれることがある。[8.2参照]
11.1.4 間質性肺疾患(1.3%)[8.3参照]
11.2 その他の副作用
| 20〜30%未満 | 10〜20%未満 | 10%未満 | |
| 胃腸障害 | 悪心 | 口内炎 | |
| 臨床検査 | 駆出率減少 | ||
| 皮膚および皮下組織障害 | 発疹 | 皮膚乾燥、そう痒症、爪の障害 |
14. 適用上の注意
14.1 薬剤交付時の注意
14.1.1 湿気を避けるため、乾燥剤を同封したボトル包装品のまま患者に交付すること。
14.1.2 湿気を避けるため、乾燥剤を同封した元の容器にて保存し、使用の都度、密栓するよう患者に指導すること。
16. 薬物動態
16.1 血中濃度
16.1.1 反復投与
HER2(ERBB2)遺伝子異常を有する進行固形癌患者24例(日本人患者6例含む)に本剤60、120、又は240mgを空腹時に1日1回15日間反復経口投与注1)したときのゾンゲルチニブの血漿中濃度の時間推移及び薬物動態パラメータは以下の図1及び表1のとおりであった1)。
図1 本剤60、120又は240mgを1日1回反復経口投与注1)したときの血漿中濃度推移(算術平均+標準偏差)
| 用量 | 投与日 | 例数 | Cmax(nmol/L) | tmax※1(h) | AUC0-24h(nmol・h/L) | t1/2(h) |
| 60mg | 1 | 4 | 1020(44.2) | 1.99(1.97-2.05) | 8240(37.0) | 9.41(17.4) |
| 15 | 4 | 1230(54.3) | 2.04(1.07-2.93) | 11600(48.4) | 9.37(15.2) | |
| 120mg | 1 | 3 | 1610(75.1) | 3.03(2.03-3.95) | 14000(61.8) | 9.51(9.87) |
| 15 | 3 | 2830(20.6) | 2.05(1.03-3.97) | 28900(23.2) | 8.52(9.39) | |
| 240mg | 1 | 17 | 3630(45.1) | 2.07(0.967-5.95) | 33900(48.9)※2 | 9.19(29.3)※2 |
| 15 | 17 | 4760(41.4) | 2.95(1.03-6.00) | 52100(43.2) | 9.58(30.1) |
16.2 吸収
16.2.1 バイオアベイラビリティ
健康成人(7例)に本剤60mg注1)を単回経口投与したときの絶対バイオアベイラビリティは76%であった2)。(外国人データ)
16.2.2 食事の影響
健康成人(16例)に本剤240mg注1)を単回経口投与したとき、空腹時投与に対する高脂肪食後投与におけるゾンゲルチニブのAUC0-t及びCmaxの幾何平均値の比は、それぞれ1.27及び1.26であった3)。(外国人データ)
16.3 分布
16.4 代謝
ゾンゲルチニブは主にCYP3A4/5による酸化、UGT1A4によるグルクロン酸抱合及びグルタチオン抱合によって代謝される。健康成人(8例)に14C標識されたゾンゲルチニブ60mg注1)を単回経口投与したとき、血漿中に未変化体及び6種類の代謝物が検出され、血漿中総放射能量(AUC0-168h)の75%が未変化体であった6)。(外国人データ)
16.5 排泄
健康成人(8例)に14C標識されたゾンゲルチニブ60mg注1)を単回経口投与したとき、投与後11日までに投与量の94%が排泄され、糞便中には93%(未変化体として31%)、尿中には1.3%(未変化体として0.2%)が排泄された7)。(外国人データ)
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
母集団薬物動態解析を用いて、腎機能注2)が正常(187例)、軽度(120例)及び中等度(23例)の腎機能障害患者にゾンゲルチニブ120mgを1日1回反復経口投与したときの曝露量を推定した結果、[1]Cmax及び[2]AUC0-24hの幾何平均値は、それぞれ[1]2730、2690及び2600nmol/L、並びに[2]30800、31700及び31200nmol・h/Lと推定された。
注2)eGFR(mL/min/1.73m2)が[1]90以上、[2]60以上90未満及び[3]30以上60未満の場合、それぞれ[1]正常、[2]軽度及び[3]中等度とされた8)。
16.6.2 肝機能障害患者
母集団薬物動態解析を用いて、肝機能注3)が正常(288例)及び軽度(90例)の肝機能障害患者にゾンゲルチニブ120mgを1日1回反復経口投与したときの曝露量を推定した結果、[1]Cmax及び[2]AUC0-24hの幾何平均値は、それぞれ[1]2700及び2630nmol/L、並びに[2]31000及び30500nmol・h/Lと推定された8)。
注3)NCI-ODWG(National Cancer Institute-Organ Dysfunction Working Group)基準による分類
16.7 薬物相互作用
16.7.1 臨床薬物相互作用試験
健康成人を対象とした臨床薬物相互作用試験から得られた、ゾンゲルチニブの薬物動態に及ぼす併用薬の影響及び併用薬の薬物動態に及ぼすゾンゲルチニブの影響は、以下の表2及び表3のとおりであった9)10)11)12)13)。[10.2参照]
| 併用薬 | 用法・用量 | 例数※1 | ゾンゲルチニブの単独投与時に対する比※2 | ||
| 併用薬 | ゾンゲルチニブ注1) | Cmax | AUC0-∞ | ||
| イトラコナゾール(強いCYP3A阻害剤) | 200mg QD | 15mg単回 | 16/16 | 1.27 (1.07,1.51) | 1.41 (1.26,1.58) |
| カルバマゼピン(強いCYP3A誘導剤) | 600mg QD※3 | 60mg単回 | 15/16 | 0.56 (0.45,0.71) | 0.36 (0.32,0.42) |
| ラベプラゾール(プロトンポンプ阻害剤) | 40mg QD | 30mg単回 | 11/12 | 0.87 (0.67,1.13) | 0.97 (0.85,1.10) |
| 併用薬 | 用法・用量 | 例数※1 | 併用薬の単独投与時に対する比※2 | ||
| 併用薬 | ゾンゲルチニブ | Cmax | AUC0-∞ | ||
| ミダゾラム (CYP3A基質) | 1mg単回 | 120mg QD | 14/16 | 1.17 (1.08,1.27) | 1.06 (0.95,1.17) |
| レパグリニド (CYP2C8基質) | 0.5mg単回 | 14/16 | 1.44 (1.24,1.69) | 1.30 (1.10,1.53) | |
| オメプラゾール (CYP2C19基質) | 20mg単回 | 13/15※3 | 0.62 (0.45,0.86) | 0.90 (0.62,1.30) | |
| ダビガトラン (P-gp基質) | 150mg単回 | 120mg単回 | 15/16 | 1.24 (0.96,1.60) | 1.34 (1.06,1.70) |
| ロスバスタチン (OATP1B1、OATP1B3、BCRP基質)※4 | 10mg単回 | 120mg QD | 16/16 | 3.02 (2.47,3.68) | 2.30 (1.96,2.71) |
| メトホルミン (MATE1、MATE2-K基質) | 10mg単回 | 16/16 | 0.76 (0.68,0.84) | 0.83 (0.76,0.90) | |
| フロセミド (OAT1、OAT3基質)※5 | 1mg単回 | 16/16 | 1.50 (1.16,1.93) | 1.38 (1.20,1.58) | |
16.7.2 その他
注1)承認された用法及び用量は、ゾンゲルチニブとして1日1回120mgである。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第Ia/Ib相試験(Beamion LUNG-1試験)16)
HER2(ERBB2)遺伝子異常を有する切除不能な進行固形癌患者を対象とした国際共同第Ia/Ib相試験の第Ib相パートにおいて、HER2(ERBB2)遺伝子変異を有する切除不能な進行・再発の非小細胞肺癌患者322例に本剤120又は240mg注)を1日1回経口投与した。
白金系抗悪性腫瘍剤を含む1つ以上の化学療法歴のあるHER2(ERBB2)遺伝子(チロシンキナーゼドメイン)に変異を有する患者を対象としたコホートにおいて、主要評価項目であるRECIST ver.1.1に基づく盲検下独立中央審査判定による奏効率[97.5%信頼区間]は、本剤120mg投与群(75例、日本人9例を含む)で66.7%[53.8,77.5](50/75例)であった(2024年5月23日データカットオフ)。
白金系抗悪性腫瘍剤を含む1つ以上の化学療法歴のあるHER2(ERBB2)遺伝子(チロシンキナーゼドメイン以外)に変異を有する患者を対象としたコホートにおいて、RECIST ver.1.1に基づく治験担当医師判定による奏効率は、本剤120mg投与群(12例)で41.7%(5/12例)であった(2024年8月29日データカットオフ)。
白金系抗悪性腫瘍剤を含む1つ以上の化学療法歴及びHER2を標的とした抗体薬物複合体による治療歴のあるHER2(ERBB2)遺伝子(チロシンキナーゼドメイン)に変異を有する患者を対象としたコホートにおいて、RECIST ver.1.1に基づく盲検下独立中央審査判定による奏効率[95%信頼区間]は、本剤120mg投与群(31例、日本人1例を含む)で41.9%[26.4,59.2](13/31例)であった(2024年8月29日データカットオフ)。
第Ib相パートで本剤120mgが投与された236例(日本人23例を含む)のうち、副作用は210例(89.0%)に認められた。主な副作用は、下痢134例(56.8%)、発疹63例(26.7%)、ALT増加43例(18.2%)、AST増加43例(18.2%)、そう痒症36例(15.3%)であった。
白金系抗悪性腫瘍剤を含む1つ以上の化学療法歴のあるHER2(ERBB2)遺伝子(チロシンキナーゼドメイン)に変異を有する患者を対象としたコホートにおいて、主要評価項目であるRECIST ver.1.1に基づく盲検下独立中央審査判定による奏効率[97.5%信頼区間]は、本剤120mg投与群(75例、日本人9例を含む)で66.7%[53.8,77.5](50/75例)であった(2024年5月23日データカットオフ)。
白金系抗悪性腫瘍剤を含む1つ以上の化学療法歴のあるHER2(ERBB2)遺伝子(チロシンキナーゼドメイン以外)に変異を有する患者を対象としたコホートにおいて、RECIST ver.1.1に基づく治験担当医師判定による奏効率は、本剤120mg投与群(12例)で41.7%(5/12例)であった(2024年8月29日データカットオフ)。
白金系抗悪性腫瘍剤を含む1つ以上の化学療法歴及びHER2を標的とした抗体薬物複合体による治療歴のあるHER2(ERBB2)遺伝子(チロシンキナーゼドメイン)に変異を有する患者を対象としたコホートにおいて、RECIST ver.1.1に基づく盲検下独立中央審査判定による奏効率[95%信頼区間]は、本剤120mg投与群(31例、日本人1例を含む)で41.9%[26.4,59.2](13/31例)であった(2024年8月29日データカットオフ)。
第Ib相パートで本剤120mgが投与された236例(日本人23例を含む)のうち、副作用は210例(89.0%)に認められた。主な副作用は、下痢134例(56.8%)、発疹63例(26.7%)、ALT増加43例(18.2%)、AST増加43例(18.2%)、そう痒症36例(15.3%)であった。
注)承認された用法及び用量は、ゾンゲルチニブとして1日1回120mgである。
18. 薬効薬理
18.1 作用機序
ゾンゲルチニブは、エクソン20挿入変異等を有するHER2のチロシンキナーゼ活性を阻害し、下流のシグナル伝達を阻害することにより、腫瘍増殖抑制作用を示すと考えられている。
18.2 抗腫瘍作用
19. 有効成分に関する理化学的知見
19.1. ゾンゲルチニブ
| 一般的名称 | ゾンゲルチニブ |
|---|---|
| 一般的名称(欧名) | Zongertinib |
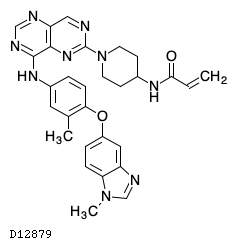
| 化学名 | N-[1-(8-{3-Methyl-4-[(1-methyl-1H-1,3-benzimidazol-5-yl)oxy]anilino}pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl]prop-2-enamide |
| 分子式 | C29H29N9O2 |
| 分子量 | 535.60g/mol |
| 物理化学的性状 | 黄色〜暗黄色又は橙色の粉末である。 |
| 分配係数 | log P=log D(pH7.4)=4.7 |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
14錠(ボトル、乾燥剤入り)
23. 主要文献
- 社内資料:国際共同第I相試験(Beamion LUNG-1)(2025年9月19日承認、CTD 2.7.2.3)
- 社内資料:ヒトADME試験(2025年9月19日承認、CTD 2.7.2.2)
- 社内資料:食事の影響試験(2025年9月19日承認、CTD 2.7.1.2)
- 社内資料:非臨床薬物動態試験(in vitro血漿タンパク結合)(2025年9月19日承認、CTD 2.6.4.4)
- 社内資料:非臨床薬物動態試験(in vitro血球移行性)(2025年9月19日承認、CTD 2.6.4.4)
- 社内資料:ヒトADME試験における代謝物同定(2025年9月19日承認、CTD 2.6.4.5)
- 社内資料:ヒトADME試験(2025年9月19日承認、CTD 2.7.2.2、CTD 2.7.2.3)
- 社内資料:母集団薬物動態解析(2025年9月19日承認、CTD 2.7.2.3)
- 社内資料:イトラコナゾールとの薬物相互作用試験(2025年9月19日承認、CTD 2.7.2.2)
- 社内資料:カルバマゼピンとの薬物相互作用試験(2025年9月19日承認、CTD 2.7.2.2)
- 社内資料:ラベプラゾールとの薬物相互作用試験(2025年9月19日承認、CTD 2.7.1.2)
- 社内資料:CYP基質とのカクテル薬物相互作用(2025年9月19日承認、CTD 2.7.2.2)
- 社内資料:トランスポーター基質とのカクテル薬物相互作用(2025年9月19日承認、CTD 2.7.2.2)
- 社内資料:非臨床薬物動態試験(in vitro薬物代謝酵素誘導及び阻害)(2025年9月19日承認、CTD 2.6.4.5)
- 社内資料:非臨床薬物動態試験(薬物トランスポーターによる輸送)(2025年9月19日承認、CTD 2.6.4.7)
- 社内資料:国際共同第I相試験(Beamion LUNG-1)
- 社内資料:非臨床薬効薬理試験(in vitro、がん細胞株における増殖阻害作用)(2025年9月19日承認、CTD 2.6.2.2)
- 社内資料:非臨床薬効薬理試験(in vivo、PDXモデルマウスにおける腫瘍増殖抑制作用)(2025年9月19日承認、CTD 2.6.2.2)
24. 文献請求先及び問い合わせ先
文献請求先
日本ベーリンガーインゲルハイム株式会社
DIセンター
〒141-6017
東京都品川区大崎2丁目1番1号 ThinkPark Tower
電話:0120-189-779 (受付時間)9:00〜18:00(土・日・祝日・弊社休業日を除く)
製品情報問い合わせ先
日本ベーリンガーインゲルハイム株式会社
DIセンター
〒141-6017
東京都品川区大崎2丁目1番1号 ThinkPark Tower
電話:0120-189-779 (受付時間)9:00〜18:00(土・日・祝日・弊社休業日を除く)
25. 保険給付上の注意
本剤は新医薬品であるため、厚生労働省告示第107号(平成18年3月6日付)に基づき、2026年11月末日までは、投薬は1回14日分を限度とされている。
26. 製造販売業者等
26.1 製造販売元
日本ベーリンガーインゲルハイム株式会社
東京都品川区大崎2丁目1番1号