医薬品情報
| 総称名 | ボラニゴ |
|---|---|
| 一般名 | ボラシデニブ クエン酸水和物 |
| 欧文一般名 | Vorasidenib Citric Acid Hydrate |
| 製剤名 | ボラシデニブ クエン酸水和物 |
| 薬効分類名 | 抗悪性腫瘍剤 |
| 薬効分類番号 | 4299 |
| KEGG DRUG |
D11835
ボラシデニブクエン酸塩水和物
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年4月 改訂(第3版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ボラニゴ錠10mg | Voranigo Tablets | 日本セルヴィエ | 4291097F1024 | 31791.8円/錠 | 劇薬, 処方箋医薬品 |
| ボラニゴ錠40mg | Voranigo Tablets | 日本セルヴィエ | 42910J1F2024 | 劇薬, 処方箋医薬品 |
1. 警告
本剤は、緊急時に十分対応できる医療施設において、がん化学療法に十分な知識・経験を持つ医師のもとで、本剤の使用が適切と判断される症例についてのみ投与すること。また、治療開始に先立ち、患者又はその家族に有効性及び危険性を十分説明し、同意を得てから投与すること。
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
IDH1又はIDH2遺伝子変異陽性の神経膠腫
5. 効能または効果に関連する注意
5.1 十分な経験を有する病理医又は検査施設における検査により、IDH1又はIDH2遺伝子変異が確認された患者に投与すること。
5.2 手術(生検術を含む)後の患者であり、直ちに放射線療法又はアルキル化剤を含む化学療法を実施する必要がない患者を対象とすること。
5.3 臨床試験に組み入れられた患者の病理組織型、組織学的悪性度、病変の画像所見等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと。[17.1.1参照]
6. 用法及び用量
通常、成人には、ボラシデニブとして40mgを1日1回、空腹時に経口投与する。
通常、12歳以上の小児には、ボラシデニブとして体重に応じて以下を1日1回、空腹時に経口投与する。
40kg未満:20mg
40kg以上:40mg
なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
7.1 他の抗悪性腫瘍剤との併用について、有効性及び安全性は確立していない。
7.2 食後に本剤を投与した場合、本剤のCmax及びAUCが増加するとの報告がある。食事の影響を避けるため、食事の1時間前から食後2時間までの間の服用は避けること。[16.2.2参照]
7.3 本剤投与中に副作用が発現した場合は、次の基準を参考に、休薬、減量又は中止の対応を行うこと。[8.、11.1.1、11.2参照]
| 副作用 | 程度* | 処置 |
| 肝機能障害 | ALT又はASTが基準値上限(ULN)の3倍超5倍以下、かつ総ビリルビン値がULNの2倍以下 | Grade 1以下又はベースラインに回復するまで休薬し、28日以内に回復した場合には、同一用量で、28日以内に回復しなかった場合は、回復後に1段階減量して再開できる。 再発した場合は、Grade 1以下又はベースラインに回復するまで休薬し、回復後に1段階減量して再開できる。 |
| ALT又はASTがULNの5倍超20倍以下、かつ総ビリルビン値がULNの2倍以下 | Grade 1以下又はベースラインに回復するまで休薬し、28日以内に回復した場合には、1段階減量して再開できる。なお、28日以内に回復しなかった場合には、投与を中止する。 再発した場合は、投与を中止する。 | |
| ALT又はASTがULNの3倍超20倍以下、かつ総ビリルビン値がULNの2倍超 ALT又はASTがULNの20倍超 | 投与を中止する。 | |
| 上記以外の副作用 | Grade 3 | Grade 1以下又はベースラインに回復するまで休薬し、回復後に1段階減量して再開できる。 再発した場合は、投与を中止する。 |
| Grade 4 | 投与を中止する。 |
| 用量調節段階 | 投与量(1日1回) | |
| 成人及び体重40kg以上の小児 | 体重40kg未満の小児 | |
| 通常投与量 | 40mg | 20mg |
| 1段階減量 | 20mg | 10mg |
| 2段階減量 | 10mg | 投与中止 |
| 3段階減量 | 投与中止 | − |
8. 重要な基本的注意
9. 特定の背景を有する患者に関する注意
9.3 肝機能障害患者
9.3.1 重度の肝機能障害患者(Child-Pugh分類C)
本剤は主に肝代謝により消失するため、血中濃度が上昇する可能性がある。なお、重度の肝機能障害患者を対象とした臨床試験は実施していない。[16.6.2参照]
9.4 生殖能を有する者
9.4.1 妊娠する可能性のある女性には、本剤投与中及び最終投与後2箇月間において避妊する必要性及び適切な避妊法について説明すること。経口避妊薬による避妊法の場合には、経口避妊薬以外の方法をあわせて使用するよう指導すること。[9.5、10.2参照]
9.4.2 男児及び生殖可能な男性に投与する場合には、造精機能の低下があらわれる可能性があることを考慮すること。ラットを用いた反復経口投与毒性試験において、臨床曝露量の24.7倍以上に相当する用量で、雄性生殖器への影響(精巣精細管変性及び萎縮、精巣上体内腔細胞残屑、前立腺及び精嚢萎縮)が認められ、精巣精細管変性は完全には回復しなかった1)。
9.5 妊婦
9.6 授乳婦
授乳しないことが望ましい。ヒトでの乳汁移行に関するデータはないが、乳汁を介して本剤を摂取した場合、乳児に重篤な副作用が発現するおそれがある。[9.5参照]
9.7 小児等
12歳未満の小児等を対象とした臨床試験は実施していない。AG881-C-004試験は12歳以上を対象として実施されたが、18歳未満の患者は本剤群に組み入れられなかった。
10. 相互作用
相互作用序文
本剤は、CYP1A2により代謝される。また、本剤はCYP2B6、2C8、2C9、2C19及び3Aに対する誘導作用並びにBCRPに対する阻害作用を有する。[16.4参照]
薬物代謝酵素用語
CYP1A2
薬物代謝酵素用語
CYP2B6
薬物代謝酵素用語
CYP2C8
薬物代謝酵素用語
CYP2C9
薬物代謝酵素用語
CYP2C19
薬物代謝酵素用語
CYP3A
薬物代謝酵素用語
BCRP
10.1 併用禁忌
10.2 併用注意
| CYP1A2阻害剤 シプロフロキサシン、ベムラフェニブ、メキシレチン等 [16.7.1参照] | 本剤の副作用が増強されるおそれがあるため、本剤の減量を考慮するとともに、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | これらの薬剤がCYP1A2を阻害することにより、本剤の血中濃度が上昇する可能性がある。 |
| CYP1A2誘導剤 フェニトイン、リファンピシン、カルバマゼピン等 [16.7.2参照] | 本剤の有効性が減弱されるおそれがあるため、CYP1A2誘導作用のない薬剤への代替を考慮すること。 | これらの薬剤がCYP1A2を誘導することにより、本剤の血中濃度が低下する可能性がある。 |
| タバコ(喫煙) | 本剤の有効性が減弱されるおそれがある。 | 喫煙によるCYP1A2の誘導により、本剤の血中濃度が低下する可能性がある。 |
| CYP3Aの基質となる薬剤 ミダゾラム、デキサメタゾン、経口避妊薬(デソゲストレル・エチニルエストラジオール、ノルエチステロン・エチニルエストラジオール、レボノルゲストレル・エチニルエストラジオール等)等 [9.4.1、16.7.2参照] | これらの薬剤の有効性が減弱するおそれがある。 | 本剤がCYP3Aを誘導することにより、これらの薬剤の血中濃度が低下する可能性がある。 |
| CYP2B6の基質となる薬剤 シクロホスファミド、エファビレンツ、メサドン等 [16.7.2参照] | これらの薬剤の有効性が減弱するおそれがある。 | 本剤がCYP2B6を誘導することにより、これらの薬剤の血中濃度が低下する可能性がある。 |
| CYP2C8の基質となる薬剤 レパグリニド、パクリタキセル、モンテルカスト等 [16.7.2参照] | これらの薬剤の有効性が減弱するおそれがある。 | 本剤がCYP2C8を誘導することにより、これらの薬剤の血中濃度が低下する可能性がある。 |
| CYP2C9の基質となる薬剤 ワルファリン、フルルビプロフェン、フェニトイン等 [16.7.2参照] | これらの薬剤の有効性が減弱するおそれがある。 | 本剤がCYP2C9を誘導することにより、これらの薬剤の血中濃度が低下する可能性がある。 |
| CYP2C19の基質となる薬剤 イホスファミド、タモキシフェンクエン酸塩、オメプラゾール等 [16.7.2参照] | これらの薬剤の有効性が減弱するおそれがある。 | 本剤がCYP2C19を誘導することにより、これらの薬剤の血中濃度が低下する可能性がある。 |
| BCRPの基質となる薬剤 ロスバスタチン、サラゾスルファピリジン、リバーロキサバン等 [16.7.2参照] | これらの薬剤の副作用が増強されるおそれがあるため、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | 本剤がBCRPを阻害することにより、これらの薬剤の血中濃度が上昇する可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には、投与を中止するなど適切な処置を行うこと。
11.1.1 肝不全(0.6%)、肝炎(自己免疫性肝炎を含む)(0.6%)、肝機能障害(40.7%)[7.3、8.参照]
11.2 その他の副作用
| 10%以上 | 5〜10%未満 | 1〜5%未満 | |
| 胃腸障害 | 悪心、下痢 | 便秘、腹痛 | 嘔吐、鼓腸、胃食道逆流性疾患、腹部膨満、上腹部痛、消化不良、おくび、胃炎 |
| 一般・全身障害 | 疲労 | 無力症 | |
| 神経系障害 | 頭痛、浮動性めまい | 注意力障害、痙攣発作 | |
| 代謝および栄養障害 | 食欲減退 | 低リン血症、高血糖、高カリウム血症 | |
| 臨床検査 | 血中乳酸脱水素酵素増加、血中尿素増加、血小板数減少 | ||
| 精神障害 | 不眠症、不安、錯乱状態 | ||
| 筋骨格系および結合組織障害 | 筋肉痛、背部痛 | ||
| 皮膚および皮下組織障害 | 脱毛症、多汗症、寝汗 | ||
| 呼吸器、胸郭および縦隔障害 | 呼吸困難 | ||
| 血管障害 | 高血圧 | ||
| 血液およびリンパ系障害 | 貧血 |
14. 適用上の注意
14.1 薬剤交付時の注意
14.1.1 本剤は吸湿性があるため、分包せずボトルで提供すること。
14.1.2 患者又は保護者等に対し以下の点に注意するよう指導すること。
・ボトル包装のふたはチャイルドロックを施しているため、ふたを押しながらねじって開封すること。
・湿気を避けるため、乾燥剤を同封した元の容器で保管すること。
・容器から乾燥剤を取り出さず、使用の都度密栓すること。
15. その他の注意
15.1 臨床使用に基づく情報
因果関係は明らかではないが、AG881-C-004試験において、本剤群の6/168例(3.6%)及びプラセボ群の2/163例(1.2%)に悪性転化*が認められた。無作為化から悪性転化までの期間の中央値[最小、最大](箇月)は、本剤群で16.6[6.9,22.5]、プラセボ群で9.2[9.0,9.4]であった(2023年3月7日データカットオフ)。
*:本剤又はプラセボの投与中止後の手術における病理組織学的検査でGrade 3又は4と確認された場合を「悪性転化」と定義した。
15.2 非臨床試験に基づく情報
ラットを用いた反復経口投与毒性試験において、臨床曝露量の24.7倍以上に相当する用量で、雌性生殖器への影響(性周期消失、卵巣黄体減少及び閉鎖卵胞増加、子宮及び腟萎縮等)が認められた。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人に本剤50mg注)を単回経口投与したときのボラシデニブの薬物動態パラメータ及び血漿中濃度推移は以下のとおりであった2)。
| Cmax(ng/mL) | AUC(hr*ng/mL) | tmax(hr) | t1/2(hr) | |
| 日本人 (15例) | 66.65 (48.78) | 3311.73 (60.50) | 2.00 (1.00,6.00) | 184.96 (40.60) |
| 外国人 (11例) | 68.29 (54.88) | 3193.13 (78.79) | 2.00 (0.50,3.00) | 212.10 (79.13) |
16.1.2 反復投与
16.2 吸収
16.3 分布
16.4 代謝
16.5 排泄
健康成人(5例)に14C標識した本剤50mg注)を単回経口投与したとき、投与1,056時間後までに投与量の84.7%が糞中、4.52%が尿中に排泄された。投与408時間後までに糞中に排泄された未変化体は投与量の55.5%であり5)、尿中に未変化体は検出されなかった(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
AG881-C-004試験の母集団薬物動態解析において、腎機能が正常な患者(eGFRが90mL/min/1.73m2以上)(210例)に対する軽度(eGFRが60mL/min/1.73m2以上90mL/min/1.73m2未満)及び中等度(eGFRが30mL/min/1.73m2以上60mL/min/1.73m2未満)の腎機能障害を有する患者(それぞれ56例及び4例)におけるボラシデニブの定常状態におけるAUCの幾何平均値の比はそれぞれ1.38及び1.26と推定された(外国人データ)。
16.6.2 肝機能障害患者
16.7 薬物相互作用
16.7.1 シプロフロキサシン
16.7.2 生理学的薬物動態モデルによるシミュレーション
(1)フルボキサミン
(2)フェニトイン、リファンピシン
本剤40mgを1日1回反復投与及びフェニトイン(中程度のCYP1A2誘導剤)又はリファンピシン(中程度のCYP1A2誘導剤)と併用投与したとき、本剤単独投与時と比較してフェニトイン又はリファンピシン併用投与時のボラシデニブの曝露量が低下する可能性が示唆された。[10.2参照]
(3)ミダゾラム、ブプロピオン、レパグリニド、ワルファリン、S-メフェニトイン
本剤40mgを1日1回反復投与及びミダゾラム(CYP3A基質)、ブプロピオン(CYP2B6基質、国内未承認)、レパグリニド(CYP2C8基質)、ワルファリン(CYP2C9基質)又はS-メフェニトイン(CYP2C19基質)と併用投与したとき、単独投与時と比較して本剤併用時にミダゾラム、ブプロピオン、レパグリニド、ワルファリン又はS-メフェニトインの曝露量が低下する可能性が示唆された。[10.2参照]
(4)ロスバスタチン
本剤40mgを1日1回反復投与及びロスバスタチン(BCRP基質)と併用投与したとき、単独投与時と比較して本剤併用時にロスバスタチンの曝露量が増加する可能性が示唆された。[10.2参照]
16.7.3 その他
(1)健康成人(21例)に、ラモトリギン(UGT1A4基質)50mgを本剤投与開始20日前及び本剤投与開始14日目に経口投与し、本剤50mg注)を1日1回15日間経口投与したとき、本剤単独投与時に対するラモトリギンの併用投与時のボラシデニブのCmax及びAUCinfの幾何平均値の比はそれぞれ0.95及び0.92であった(外国人データ)。
(2)健康成人(35例)に、オメプラゾール(プロトンポンプ阻害剤)40mgを1日1回4日間経口投与し、本剤50mg注)を単回経口投与したとき、本剤単独投与時に対するオメプラゾールの併用投与時のボラシデニブのCmax及びAUCinfの幾何平均値の比はそれぞれ0.72及び0.94であった(外国人データ)。
注)
本剤の承認された用法及び用量は、「通常、成人には、ボラシデニブとして40mgを1日1回、空腹時に経口投与する。
通常、12歳以上の小児には、ボラシデニブとして体重に応じて以下を1日1回、空腹時に経口投与する。
40kg未満:20mg
40kg以上:40mg
なお、患者の状態により適宜減量する。」である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第III相試験(AG881-C-004試験)
12歳以上かつ体重40kg以上の、手術歴があり注1)、放射線療法又は化学療法による治療歴のないIDH1又はIDH2遺伝子変異陽性注2)のグレード2注3)の残存又は再発の星細胞腫及び乏突起膠腫患者注4)(直ちに放射線療法又はアルキル化剤を含む化学療法を行う必要のある患者を除く注5))を対象に、無作為化二重盲検プラセボ対照試験を実施した。疾患進行又は治験中止基準に該当するまで、本剤40mg又はプラセボを1日1回経口投与した。主要評価項目は改訂Response Assessment in Neuro-Oncology for Low Grade Glioma(RANO-LGG)基準に基づく盲検下独立評価委員会による無増悪生存期間(PFS)と設定された。中間解析時点(2022年9月6日データカットオフ)におけるPFSの結果は、以下のとおりであった注6)。
注1)直近の手術(生検を含む)が無作為化より1年以上前(IDH遺伝子検査を目的とした生検を除く)かつ5年以内の患者が対象とされた。
注2)中央検査機関による検査によりIDH1 R132H/C/G/S/L変異又はIDH2 R172K/M/W/S/G変異を有することが確認された患者が対象とされた。
注3)WHO 2016脳腫瘍分類に基づく。
注4)非造影病変(MRI評価において造影剤で増強されない病変)を有する患者が組み入れられた。造影病変を有する患者は、直近2回のMRI評価で病変に変化がなく、中央判定により最小、非結節性かつ測定不能病変であることが確認された場合にのみ組入れ可能とされた。
注5)脳幹浸潤病変、腫瘍による臨床的に重要な機能的障害又は神経認知障害(手術に起因する障害を除く)、コントロール不良の痙攣発作(日常生活活動に支障を来す持続性の発作を有し、かつ抗てんかん薬による3つ以上の治療(うち、少なくとも1つは抗てんかん薬の併用療法)に不応である発作)等、治験責任医師の評価に基づき高リスクの特徴を有する患者は除外することとされた。
注6)データカットオフ時点において、日本人患者は組み入れられていなかった。
| 本剤群 (168例) | プラセボ群 (163例) | |
| イベント例数(%) | 47(28) | 88(54) |
| 中央値[月] (95%信頼区間) | 27.7(17.0,NE) | 11.1(11.0,13.7) |
| ハザード比注7) (95%信頼区間) | 0.39(0.27,0.56)注8) | |
| P値注9) | <0.0001 | |
無増悪生存期間のKaplan-Meier曲線(AG881-C-004試験)
中間解析のデータカットオフ(2022年9月6日)後に組み入れられた日本人部分集団16例注10)において、2024年3月14日データカットオフ時点でPFS中央値は未達であり、イベントは認められなかった(追跡調査期間の中央値[95%信頼区間](箇月):11.1[8.4,11.1])。
中間解析時点(2022年9月6日データカットオフ)における副作用発現頻度は、本剤群で65.3%(109/167例)であった。主な副作用(10%以上)は、ALT増加36.5%(61/167例)、AST増加24.6%(41/167例)、疲労21.0%(35/167例)、悪心15.0%(25/167例)、GGT増加13.2%(22/167例)、下痢12.0%(20/167例)であった。
日本人部分集団における2024年3月14日データカットオフ時点の副作用発現頻度は、81.3%(13/16例)であった。主な副作用(10%以上)は、ALT増加68.8%(11/16例)、AST増加56.3%(9/16例)、GGT増加31.3%(5/16例)、悪心12.5%(2/16例)であった。[5.3参照]
中間解析時点(2022年9月6日データカットオフ)における副作用発現頻度は、本剤群で65.3%(109/167例)であった。主な副作用(10%以上)は、ALT増加36.5%(61/167例)、AST増加24.6%(41/167例)、疲労21.0%(35/167例)、悪心15.0%(25/167例)、GGT増加13.2%(22/167例)、下痢12.0%(20/167例)であった。
日本人部分集団における2024年3月14日データカットオフ時点の副作用発現頻度は、81.3%(13/16例)であった。主な副作用(10%以上)は、ALT増加68.8%(11/16例)、AST増加56.3%(9/16例)、GGT増加31.3%(5/16例)、悪心12.5%(2/16例)であった。[5.3参照]
注10)プラセボ群への割り付け後、本剤群へクロスオーバーした9例を含む。
18. 薬効薬理
18.1 作用機序
ボラシデニブは、イソクエン酸脱水素酵素(IDH)1及びIDH2に対する阻害作用を有する低分子化合物である。ボラシデニブは、変異型IDH1及びIDH2の酵素活性を阻害することで腫瘍細胞における2-ヒドロキシグルタル酸(2-HG)産生を阻害し、IDH1又はIDH2遺伝子変異陽性の腫瘍細胞の分化を誘導すること等により、腫瘍増殖抑制作用を示すと考えられている。
18.2 2-HG産生阻害作用
ボラシデニブは、変異型IDH1(R132H)を発現するヒト神経膠腫由来TS603細胞株を皮下移植した重症複合型免疫不全マウス及び変異型IDH2(R140Q)を発現させたヒト神経膠腫由来U87MG細胞株を同所移植したヌードマウスにおいて、2-HG産生を阻害した(in vivo)。
19. 有効成分に関する理化学的知見
19.1. ボラシデニブ クエン酸水和物
| 一般的名称 | ボラシデニブ クエン酸水和物 |
|---|---|
| 一般的名称(欧名) | Vorasidenib Citric Acid Hydrate |
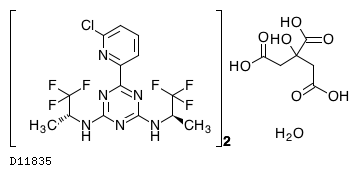
| 化学名 | 6-(6-Chloropyridin-2-yl)-N2,N4-bis[(2R)-1,1,1-trifluoropropan-2-yl]-1,3,5-triazine-2,4-diamine hemicitric acid hemihydrate |
| 分子式 | (C14H13ClF6N6)2・C6H8O7・H2O |
| 分子量 | 1,039.61 |
| 融点 | 160〜165℃ |
| 物理化学的性状 | 白色の粉末である。 |
| 溶解性 | アセトン、アセトニトリル又はエタノールにやや溶けにくく、ヘプタン又は水にほとんど溶けない。 |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
<ボラニゴ錠10mg>
30錠(プラスチックボトル、バラ、乾燥剤入り)
<ボラニゴ錠40mg>
30錠(プラスチックボトル、バラ、乾燥剤入り)
薬価未収載品
23. 主要文献
- 社内資料:非臨床試験(CTD2.6.6.3.1.5)
- 社内資料:海外第I相(AG881-C-008)試験(CTD2.7.2.2.2.1)
- 社内資料:国際共同第III相(AG881-C-004)試験(CTD2.7.2.3.1)
- 社内資料:海外第I相(AG881-C-002)試験(CTD2.7.2.2.2.2)
- 社内資料:海外第I相(AG881-C-005)試験(CTD2.7.2.2.2.1)
- 社内資料:海外第I相(AG881-C-007)試験(CTD2.7.2.2.2.1)
- 社内資料:海外第I相(PKH-95032-009)試験(CTD2.7.2.2.2.1)
- 社内資料:非臨床試験(CTD2.6.4.4.3)
- 社内資料:海外第I相(AG120-881-C-001)試験(CTD2.7.2.2.2.2)
- 社内資料:海外第I相(AG881-C-005)試験(CTD2.7.2.2.2.1)
- 社内資料:海外第I相(PKH-95032-008)試験(CTD2.7.2.2.2.1)
24. 文献請求先及び問い合わせ先
文献請求先
日本セルヴィエ株式会社
お問い合わせ窓口
〒100-0004
東京都千代田区大手町1-9-2 大手町フィナンシャルシティグランキューブ
電話:0120-841-002 月〜金 9:00-17:00(祝日、弊社休業日を除く)
製品情報問い合わせ先
日本セルヴィエ株式会社
お問い合わせ窓口
〒100-0004
東京都千代田区大手町1-9-2 大手町フィナンシャルシティグランキューブ
電話:0120-841-002 月〜金 9:00-17:00(祝日、弊社休業日を除く)
25. 保険給付上の注意
本剤10mg錠は、厚生労働省告示第91号(令和8年3月17日付)に基づき、2027年3月末日までは、投薬は1回30日分を限度とされている。
26. 製造販売業者等
26.1 製造販売元
日本セルヴィエ株式会社
東京都千代田区大手町1-9-2 大手町フィナンシャルシティグランキューブ