医薬品情報
| 総称名 | ネクサバール |
|---|---|
| 一般名 | ソラフェニブトシル酸塩 |
| 欧文一般名 | Sorafenib Tosilate |
| 製剤名 | ソラフェニブトシル酸塩錠 |
| 薬効分類名 | 抗悪性腫瘍剤 キナーゼ阻害剤 |
| 薬効分類番号 | 4291 |
| ATCコード | L01EX02 |
| KEGG DRUG |
D06272
ソラフェニブトシル酸塩
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年4月 改訂(第8版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ネクサバール錠200mg | Nexavar tablets 200mg | バイエル薬品 | 4291017F1025 | 2471円/錠 | 劇薬, 処方箋医薬品 |
1. 警告
本剤は、緊急時に十分対応できる医療施設において、がん化学療法に十分な知識・経験を持つ医師のもとで、本剤の投与が適切と判断される症例についてのみ投与すること。また、治療開始に先立ち、患者又はその家族に本剤の有効性及び危険性を十分説明し、同意を得てから投与すること。
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し重篤な過敏症の既往歴のある患者
2.2 妊婦又は妊娠している可能性のある女性[9.5参照]
4. 効能または効果
5. 効能または効果に関連する注意
<根治切除不能又は転移性の腎細胞癌>
5.2 本剤の術後補助化学療法における有効性及び安全性は確立していない。
<切除不能な肝細胞癌>
5.3 局所療法(経皮的エタノール注入療法、ラジオ波焼灼療法、マイクロ波凝固療法、肝動脈塞栓療法/肝動脈化学塞栓療法、放射線療法等)の適応となる肝細胞癌患者に対する本剤の有効性及び安全性は確立していない。
5.4 肝細胞癌に対する切除及び局所療法後の補助化学療法における本剤の有効性及び安全性は確立していない。
5.5 肝機能障害の程度、局所療法の適応の有無、全身化学療法歴等について、「17.臨床成績」の項の内容に準じて、適応患者の選択を行うこと。
<根治切除不能な甲状腺癌>
5.6 臨床試験に組み入れられた患者の病理組織型等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分理解した上で、適応患者の選択を行うこと。
5.7 甲状腺未分化癌患者に対する本剤の有効性及び安全性は確立していない。
5.8 放射性ヨウ素による治療歴のない分化型甲状腺癌患者に対する本剤の有効性及び安全性は確立していない。
6. 用法及び用量
通常、成人にはソラフェニブとして1回400mgを1日2回経口投与する。なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
<効能共通>
7.2 高脂肪食の食後に本剤を投与した場合、血漿中濃度が低下するとの報告がある。高脂肪食摂取時には食事の1時間前から食後2時間までの間を避けて服用すること。[16.2.1参照]
<切除不能な肝細胞癌>
7.3 肝細胞癌に対する局所療法との併用について、有効性及び安全性は確立していない。
<根治切除不能又は転移性の腎細胞癌、切除不能な肝細胞癌>
7.4 副作用により本剤を減量、休薬又は中止する場合には、副作用の症状、重症度等に応じて以下の基準を考慮すること。
| 用量調節段階 | 投与量 |
| 通常投与量 | 1回400mgを1日2回経口投与 |
| 1段階減量 | 1回400mgを1日1回経口投与 |
| 2段階減量 | 1回400mgを隔日経口投与 |
| 皮膚の副作用のグレード | 発現回数 | 投与量の調節 |
| グレード1:手足の皮膚の感覚障害、刺痛、痛みを伴わない腫脹や紅斑、日常生活に支障を来さない程度の不快な症状 | 回数問わず | 本剤の投与を継続し、症状緩和のための局所療法を考慮する。 |
| グレード2:手足の皮膚の痛みを伴う紅斑や腫脹、日常生活に支障を来す不快な症状 | 1回目 | 本剤の投与を継続し、症状緩和のための局所療法を考慮する。 7日以内に改善が見られない場合は下記参照。 |
| 7日以内に改善が見られない場合 あるいは 2回目又は3回目 | グレード0〜1に軽快するまで休薬する。 本剤の投与を再開する場合は投与量を1段階下げる。(400mg1日1回又は400mg隔日1回) | |
| 4回目 | 本剤の投与を中止する。 | |
| グレード3:手足の皮膚の湿性落屑、潰瘍形成、水疱形成、激しい痛み、仕事や日常生活が不可能になる重度の不快な症状 | 1回目又は2回目 | グレード0〜1に軽快するまで休薬する。 本剤の投与を再開する場合は投与量を1段階下げる。(400mg1日1回又は400mg隔日1回) |
| 3回目 | 本剤の投与を中止する。 |
| グレード | 投与継続の可否 | 用量調節 |
| グレード0〜2 | 投与継続 | 変更なし |
| グレード3 | グレード0〜2に軽快するまで休薬b | 1段階下げるc |
| グレード4 | 投与中止 | 投与中止 |
<根治切除不能な甲状腺癌>
7.5 副作用により本剤を減量、休薬又は中止する場合には、副作用の症状、重症度等に応じて以下の基準を考慮すること。
| 用量調節段階 | 投与量 |
| 通常投与量 | 1回400mgを1日2回経口投与 |
| 1段階減量 | 1回400mgと1回200mgとを交互に12時間間隔で経口投与 |
| 2段階減量 | 1回200mgを1日2回経口投与 |
| 3段階減量 | 1回200mgを1日1回経口投与 |
| 皮膚の副作用のグレード | 発現回数 | 投与量の調節a |
| グレード1:手足の皮膚の感覚障害、刺痛、痛みを伴わない腫脹や紅斑、日常生活に支障を来さない程度の不快な症状 | 回数問わず | 本剤の投与を継続し、症状緩和のための局所療法を考慮する。 |
| グレード2:手足の皮膚の痛みを伴う紅斑や腫脹、日常生活に支障を来す不快な症状 | 1回目 | 本剤の投与を継続し、症状緩和のための局所療法及び1段階減量を考慮する。 7日以内に改善が見られない場合は下記参照。 |
| 7日以内に改善が見られない場合又は2回目 | グレード0〜1に軽快するまで休薬する。 本剤の投与を再開する場合は投与量を1段階下げる。 | |
| 3回目 | グレード0〜1に軽快するまで休薬する。 本剤の投与を再開する場合は投与量を2段階下げる。b | |
| 4回目 | 本剤の投与を中止する。 | |
| グレード3:手足の皮膚の湿性落屑、潰瘍形成、水疱形成、激しい痛み、仕事や日常生活が不可能になる重度の不快な症状 | 1回目 | グレード0〜1に軽快するまで休薬する。 本剤の投与を再開する場合は投与量を1段階下げる。 |
| 2回目 | グレード0〜1に軽快するまで休薬する。 本剤の投与を再開する場合は投与量を2段階下げる。 | |
| 3回目 | 本剤の投与を中止する。 |
| グレード | 発現回数 | 投与継続の可否 | 用量調節 |
| グレード0〜1 | 回数問わず | 投与継続 | 変更なし |
| グレード2 | 回数問わず | 投与継続 | 1段階下げるc、d |
| グレード3 | 1回目 | グレード0〜2に軽快するまで休薬b 7日以内に改善が見られない場合は下記参照。 | 1段階下げるc、d |
| 7日以内に改善が見られない場合 あるいは 2回目又は3回目 | グレード0〜2に軽快するまで休薬b | 2段階下げるc、d | |
| 4回目 | グレード0〜2に軽快するまで休薬b | 3段階下げるc、d | |
| グレード4 | 回数問わず | 投与中止 | 投与中止 |
8. 重要な基本的注意
<効能共通>
8.1 手足症候群、はく脱性皮膚炎、中毒性表皮壊死融解症(Toxic Epidermal Necrolysis:TEN)、皮膚粘膜眼症候群(Stevens-Johnson症候群)、多形紅斑、ケラトアカントーマ、皮膚有棘細胞癌があらわれることがあるので、必要に応じて皮膚科を受診するよう、患者に指導すること。[7.4、7.5、11.1.1-11.1.3参照]
8.2 肝機能障害、黄疸、肝不全があらわれることがあるので、本剤投与中は定期的に肝機能検査を行い、患者の状態を十分に観察すること。
なお、主に肝細胞癌又は肝硬変のある患者において肝性脳症が報告されているので、これらの患者に投与する際は、血中アンモニア値等の検査を行うとともに、意識障害等の臨床症状を十分に観察すること。[11.1.5参照]
なお、主に肝細胞癌又は肝硬変のある患者において肝性脳症が報告されているので、これらの患者に投与する際は、血中アンモニア値等の検査を行うとともに、意識障害等の臨床症状を十分に観察すること。[11.1.5参照]
8.3 急性肺障害、間質性肺炎があらわれることがあるので、呼吸困難、発熱、咳嗽等の臨床症状を十分に観察すること。また、呼吸困難、発熱、咳嗽等の症状があらわれた場合には速やかに連絡するよう患者に説明すること。[11.1.6参照]
8.4 血圧の上昇が認められることがあるので、本剤投与中は定期的に血圧測定を行うことが望ましい。高血圧クリーゼがあらわれることがあるので、血圧の推移等に十分注意しながら投与すること。高血圧があらわれた場合には、降圧剤の投与など適切な処置を行うこと。重症、持続性あるいは通常の降圧治療でコントロールできない高血圧があらわれた場合には、投与の中止を考慮すること。[9.1.1、11.1.7参照]
8.5 白血球減少、好中球減少、リンパ球減少、血小板減少、貧血があらわれることがあるので、定期的に白血球分画を含む血液学的検査を行うなど、患者の状態を十分に観察すること。[7.4、7.5、11.1.13参照]
8.6 血清アミラーゼや血清リパーゼの上昇があらわれることがあるので、本剤投与中は定期的に膵酵素を含む血液検査を行うこと。[11.1.14参照]
8.7 創傷治癒を遅らせる可能性があるので、手術時は投与を中断することが望ましい。手術後の投与再開は患者の状態に応じて判断すること。
8.8 腫瘍崩壊症候群があらわれることがあるので、血清中電解質濃度及び腎機能検査を行うなど、患者の状態を十分に観察すること。[11.1.22参照]
<根治切除不能な甲状腺癌>
8.9 定期的に血清カルシウム濃度を測定すること。
8.10 定期的に甲状腺刺激ホルモン濃度を測定すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 高血圧症の患者
9.1.2 血栓塞栓症の既往のある患者
心筋虚血、心筋梗塞などがあらわれるおそれがある。[11.1.9参照]
9.1.3 脳転移のある患者
脳出血があらわれるおそれがある。
9.3 肝機能障害患者
9.3.1 重度の肝機能障害(Child-Pugh分類C)のある患者
臨床試験で除外されている。[16.6.2参照]
9.4 生殖能を有する者
妊娠可能な女性に対しては、投与中及び投与中止後少なくとも2週間は有効な避妊を行うよう指導すること。[9.5参照]
9.5 妊婦
9.6 授乳婦
授乳しないことが望ましい。動物実験(ラット、経口投与)で乳汁中へ移行することが報告されている。
9.7 小児等
小児等を対象とした臨床試験は実施していない。動物実験で成長段階の若齢イヌに骨及び歯への影響が報告されている3)。
9.8 高齢者
患者の状態を観察しながら慎重に投与すること。一般に高齢者では生理機能が低下していることが多い。
10. 相互作用
相互作用序文
In vitro試験において、本剤は薬物代謝酵素チトクロームP450 3A4(CYP3A4)による酸化的代謝とグルクロン酸転移酵素(UGT1A9)によるグルクロン酸抱合により代謝されることが示されているので、本酵素の活性に影響を及ぼす薬剤と併用する場合には、注意して投与すること。また、in vitro試験で、本剤のUGT1A1、UGT1A9、CYP2B6、CYP2C9及びCYP2C8に対する阻害活性が示されており、これらの酵素により代謝される他の薬剤の血中濃度を上昇させる可能性がある。
薬物代謝酵素用語
CYP3A4
薬物代謝酵素用語
グルクロン酸転移酵素(UGT1A9)
薬物代謝酵素用語
UGT1A1
薬物代謝酵素用語
CYP2B6
薬物代謝酵素用語
CYP2C9
薬物代謝酵素用語
CYP2C8
10.2 併用注意
| イリノテカン | イリノテカン及びその活性代謝物であるSN-38のAUCがそれぞれ26〜42%及び67〜120%増加するとの報告がある4)。 | 本剤はUGT1A1によるグルクロン酸抱合を阻害することにより、SN-38の代謝を阻害し、血中濃度を上昇させる可能性がある。 |
| ドキソルビシン | ドキソルビシンのAUCが21%増加したとの報告がある5)。 | 機序不明 |
| CYP3A4誘導薬(リファンピシン、フェノバルビタール、フェニトイン、カルバマゼピン、デキサメタゾン等)及びセイヨウオトギリソウ(セント・ジョーンズ・ワート)含有食品 | リファンピシンとの併用により本剤のAUCが37%減少したとの報告がある6)。 CYP3A4誘導薬等の併用により本剤の血漿中濃度が低下する可能性がある。 | In vitro試験において、本剤はCYP3A4によって代謝されることが示唆されている。 |
| ワルファリン [16.7.1参照] | ワルファリンを併用した症例において、出血又はプロトロンビン時間の延長(INR値の上昇)の報告がある7)。 本剤とワルファリンを併用する場合には、定期的にプロトロンビン時間又はINRのモニタリングを行うこと。 | 機序不明 |
| ドセタキセル | ドセタキセルのAUCが36〜80%増加したとの報告がある8)。 | 機序不明 |
| パクリタキセル/カルボプラチン | パクリタキセル及びカルボプラチンとの併用により本剤のAUCが47%増加し、パクリタキセル及びその活性代謝物である6-OHパクリタキセルのAUCがそれぞれ29%及び50%増加したとの報告がある。 | 機序不明 |
| カペシタビン | カペシタビン及びその活性代謝物であるフルオロウラシルのAUCがそれぞれ50%及び52%増加したとの報告がある。 | 機序不明 |
| フラジオマイシン(経口剤:国内未発売) [16.7.2参照] | フラジオマイシンとの併用により本剤のAUCが54%低下したとの報告がある9)。 抗生物質との併用により本剤の血漿中濃度が低下する可能性がある。 | フラジオマイシンの腸内細菌叢への影響により、本剤の腸肝循環が抑制される。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 手足症候群(46.7%)、はく脱性皮膚炎(頻度不明)
11.1.2 中毒性表皮壊死融解症(Toxic Epidermal Necrolysis:TEN)(頻度不明)、皮膚粘膜眼症候群(Stevens-Johnson症候群)(頻度不明)、多形紅斑(1.4%)[8.1参照]
11.1.3 ケラトアカントーマ(0.6%)、皮膚有棘細胞癌(0.6%)[8.1参照]
11.1.4 出血(消化管出血、気道出血、脳出血、口腔内出血、鼻出血、爪床出血、血腫、腫瘍出血)(7.5%)
死亡に至る例が報告されている。重篤な出血が認められた場合には投与を中止し、適切な処置を行うこと。
11.1.5 劇症肝炎(頻度不明)、肝機能障害・黄疸(0.8%)、肝不全(頻度不明)、肝性脳症(頻度不明)
劇症肝炎、AST、ALTの上昇を伴う肝機能障害、黄疸、肝不全、肝性脳症があらわれることがある。[8.2参照]
11.1.6 急性肺障害、間質性肺炎(いずれも頻度不明)
異常が認められた場合には速やかに胸部X線検査等を実施すること。急性肺障害、間質性肺炎が疑われた場合には投与を中止し、副腎皮質ホルモン剤の投与等の適切な処置を行うこと。[8.3参照]
11.1.8 可逆性後白質脳症症候群(頻度不明)
可逆性後白質脳症症候群が疑われた場合は、本剤の投与を中止し、血圧のコントロール、抗痙攣薬の投与等の適切な処置を行うこと。
11.1.9 心筋虚血・心筋梗塞(1.1%)
死亡に至る例が報告されている。[9.1.2参照]
11.1.10 うっ血性心不全(0.3%)
死亡に至る例が報告されている。
11.1.11 消化管穿孔(頻度不明)、消化管潰瘍(0.3%)
消化管穿孔により死亡に至る例が報告されている。
11.1.12 出血性腸炎、虚血性腸炎(いずれも頻度不明)
出血性腸炎、虚血性腸炎等の重篤な腸炎があらわれることがある。激しい腹痛・下痢・血便等の症状があらわれた場合には投与を中止し、適切な処置を行うこと。
11.1.13 白血球減少(1.5%)、好中球減少(1.2%)、リンパ球減少(1.8%)、血小板減少(2.1%)、貧血(3.4%)
11.1.14 膵炎(0.3%)
腹痛等の膵炎を示唆する症状が認められた場合や膵酵素上昇が持続する場合には、本剤を休薬又は投与中止し、適切な処置を行うこと。[8.6参照]
11.1.15 腎不全(頻度不明)
11.1.16 ネフローゼ症候群(頻度不明)、タンパク尿(1.8%)
11.1.17 低ナトリウム血症(0.6%)
意識障害、全身倦怠感、嘔吐等を伴う低ナトリウム血症があらわれることがある。
11.1.18 ショック、アナフィラキシー(いずれも頻度不明)
呼吸困難、血管性浮腫、発疹、血圧低下等があらわれることがある。
11.1.19 横紋筋融解症(頻度不明)
筋肉痛、脱力感、CK上昇、血中及び尿中ミオグロビン上昇等が認められた場合には、投与を中止し、適切な処置を行うこと。また、横紋筋融解症による急性腎障害の発症に注意すること。
11.1.20 低カルシウム血症(2.8%)
異常が認められた場合には、血清カルシウム濃度を確認し、カルシウム剤やビタミンD製剤の投与等の適切な処置を行うこと。
11.1.21 動脈解離(頻度不明)
大動脈解離を含む動脈解離があらわれることがある10)。
11.1.22 腫瘍崩壊症候群(頻度不明)
異常が認められた場合には投与を中止し、適切な処置(生理食塩液、高尿酸血症治療剤等の投与、透析等)を行うとともに、症状が回復するまで患者の状態を十分に観察すること。[8.8参照]
11.2 その他の副作用
| 10%以上 | 1〜10%未満 | 0.1〜1%未満 | 頻度不明 | |
| 過敏症 | 過敏性反応(皮膚反応及びじん麻疹を含む) | |||
| 血液 | プロトロンビン時間延長、INR上昇 | |||
| 皮膚 | 脱毛、発疹・皮膚落屑、そう痒 | 皮膚乾燥、潮紅、ざ瘡 | 白血球破砕性血管炎、紅斑、過角化、湿疹 | |
| 精神神経系 | 末梢感覚神経障害、浮動性めまい | うつ、耳鳴 | ||
| 筋・骨格系 | 関節痛、筋痛 | 筋痙縮 | ||
| 呼吸器 | 嗄声 | 鼻漏 | ||
| 循環器 | 高血圧 | QT延長 | ||
| 消化器 | 下痢、リパーゼ上昇、口内炎(口内乾燥及び舌痛を含む)、食欲不振、悪心 | アミラーゼ上昇、便秘、嘔吐、消化不良、嚥下障害 | 胃炎 | 胃食道逆流性疾患 |
| 肝臓 | ALT上昇、AST上昇、Al-P上昇、ビリルビン上昇 | 胆のう炎 | 胆管炎、LDH上昇 | |
| その他 | 疼痛(口内疼痛、腹痛、骨痛、頭痛及びがん疼痛を含む)、疲労、体重減少 | 発熱、感染、浮腫、味覚異常、粘膜の炎症、低リン酸血症、低カリウム血症、インフルエンザ様症状、無力症、脱水 | 甲状腺機能低下、甲状腺機能亢進、高カリウム血症、勃起不全、女性化乳房 | 放射線照射リコール反応、毛包炎 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.2 非臨床試験に基づく情報
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
日本人固形癌患者6例に本剤400mgを単回経口投与した際の血漿中濃度は、投与8時間後に最高血漿中濃度(Cmax)1.21mg/Lに達した。消失半減期(t1/2)は25.5時間であり、血漿中濃度時間曲線下面積(AUC)は35.4mg・h/Lであった14)。
| 投与量 | AUC(mg・h/L) | Cmax(mg/L) | tmax※(h) | t1/2(h) | CL/f(L/h) |
| 400mg | 35.4(3.50) | 1.21(3.57) | 8[3〜24] | 25.5(1.47) | 11.3(3.50) |
16.1.2 反復投与
日本人固形癌患者6例に本剤400mgを1日2回反復投与した際、投与開始10日後には定常状態に達した。定常状態における血漿中濃度推移は平坦であり、一定の濃度を維持していた。反復投与開始から14日後のCmax及びAUCは、それぞれ、4.9mg/L及び36.7mg・h/Lであった14)。
| 投与量 | AUC(0-12)(mg・h/L) | Cmax(mg/L) |
| 400mg | 36.7(1.92) | 4.9(1.96) |
16.2 吸収
16.2.1 食事の影響
16.3 分布
本剤は血漿タンパクと高い結合能を示し、ヒト血漿タンパク結合率は99.5%であった。主にアルブミンと結合し、その他にα-グロブリン、β-グロブリン及び低比重リポタンパク(LDL)にも結合した16)(in vitro試験)。
16.4 代謝
16.5 排泄
健康成人4例に[14C]ソラフェニブ100mgを溶液にて単回経口投与した場合、投与14日目までに糞中に77%、尿中に19%が回収され合計96%が排泄された。糞中には未変化体として50.7%が、カルボン酸体として19.1%が排泄された。尿中にはソラフェニブのグルクロン酸抱合体が14.8%、ピリジン基のN-酸化体のグルクロン酸抱合体が2.7%排泄された17)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
軽度の腎機能障害(クレアチニンクリアランス(Ccr)50〜80mL/min)、中等度の腎機能障害(Ccr30〜<50mL/min)及び、重度の腎機能障害(Ccr<30mL/min)を有する被験者に、本剤400mgを経口投与した場合、腎機能低下による本剤の薬物動態への影響は見られなかった18)(外国人データ)。
なお、腎透析を受けている患者における検討は行っていない。
なお、腎透析を受けている患者における検討は行っていない。
16.6.2 肝機能障害患者
16.7 薬物相互作用
16.7.1 ワルファリン
16.7.2 フラジオマイシン
本剤の血中薬物動態に対する腸肝循環の寄与を評価するため、健康成人にフラジオマイシン(経口剤:国内未発売)を5日間反復経口投与して腸内細菌叢を死滅させたのち、本剤を経口投与したところ、本剤のAUCが54%減少した9)(外国人データ)。[10.2参照]
フラジオマイシン以外の抗生物質との相互作用については検討していない。
16.7.3 その他の薬剤
本剤はCYP3A4及びUGT1A9により代謝されるが、健康成人16例に、ケトコナゾール(400mg)を1日1回7日間反復投与中に、本剤50mgを単回経口投与した際のAUCに変化は認められなかった21)(外国人データ)。
本剤はCYP2C19、2D6及び3A4をKi値17、22及び29μMで阻害したが20)(in vitro試験)、それぞれのプローブ基質であるミダゾラム、デキストロメトルファン及びオメプラゾールを本剤400mg1日2回28日間反復投与時に併用しても、これら薬物の曝露量に変化は認められなかった22)(外国人データ)。
本剤は、CYP2B6及びCYP2C8をKi値5〜6μM及び1〜2μMで阻害した20)(in vitro試験)。
17. 臨床成績
17.1 有効性及び安全性に関する試験
<根治切除不能又は転移性の腎細胞癌>
17.1.1 国内第II相試験
サイトカイン製剤(インターフェロンα、インターフェロンγ、インターロイキン2)及び腎摘出の治療歴のある切除不能又は転移性腎細胞癌患者を対象として、非対照、非盲検により、RECISTによる奏効率を有効性の主要評価項目とする試験を実施した。
有効性評価対象例129例のうち、組織型分類では淡明細胞癌が112例(86.8%)であり、Motzerリスク分類では低リスク患者が52例(40.3%)、中等度リスク患者が77例(59.7%)であった。腫瘍評価判定委員会により16例が奏効と判定された(奏効率:12.4%[95%信頼区間:7.3〜19.4%])。
安全性評価対象131例中127例(96.9%)に副作用が認められた。主な副作用は、リパーゼ増加73例(55.7%)、手足皮膚反応72例(55.0%)、脱毛51例(38.9%)、アミラーゼ増加50例(38.2%)、皮疹49例(37.4%)、下痢44例(33.6%)、代謝/検査−その他39例(29.8%)、高血圧36例(27.5%)であった23)。[5.1、7.1参照]
有効性評価対象例129例のうち、組織型分類では淡明細胞癌が112例(86.8%)であり、Motzerリスク分類では低リスク患者が52例(40.3%)、中等度リスク患者が77例(59.7%)であった。腫瘍評価判定委員会により16例が奏効と判定された(奏効率:12.4%[95%信頼区間:7.3〜19.4%])。
安全性評価対象131例中127例(96.9%)に副作用が認められた。主な副作用は、リパーゼ増加73例(55.7%)、手足皮膚反応72例(55.0%)、脱毛51例(38.9%)、アミラーゼ増加50例(38.2%)、皮疹49例(37.4%)、下痢44例(33.6%)、代謝/検査−その他39例(29.8%)、高血圧36例(27.5%)であった23)。[5.1、7.1参照]
17.1.2 海外第III相試験
全身投与による治療1レジメン(インターフェロンα、インターロイキン2等)の治療歴がある切除不能又は転移性腎細胞癌患者を対象として、プラセボ対照、無作為化、二重盲検により、全生存期間(OS)を主要評価項目、無増悪生存期間(PFS)、奏効率等を副次的評価項目とする試験を実施した。
有効性評価対象となったのは、769例(ソラフェニブ群384例、プラセボ群385例)であり、組織型分類では、ソラフェニブ群377例(98.2%)、プラセボ群380例(98.7%)が淡明細胞癌であった。Motzerリスク分類では、ソラフェニブ群の200例(52.1%)、プラセボ群の194例(50.4%)が低リスク患者であり、他は中等度リスク患者であった。有効性評価対象例において、PFSの中央値はプラセボ群で84日、ソラフェニブ群で168日であった。PFSに関する解析はMotzerリスク分類及び国による層別Log-rank検定により行い、その結果、ソラフェニブのPFSに対する効果は有意であった(p<0.000001)。ハザード比(ソラフェニブ/プラセボ)は0.51(95%信頼区間:0.43〜0.60)であった。また、OSについて、イベント(死亡)数が220にて中間解析を行った結果、層別Log-rank検定のp値は0.015であり、中間解析の有意水準として設定された0.0005には至らなかったものの、ハザード比(ソラフェニブ/プラセボ)は0.71(95%信頼区間:0.54〜0.94)であり、ソラフェニブ群ではプラセボ群に比して39%の延長を示した24)。
ソラフェニブ群の安全性評価対象384例中282例(73.4%)に副作用が認められた。主な副作用は、皮疹120例(31.3%)、下痢116例(30.2%)、手足皮膚反応101例(26.3%)、脱毛87例(22.7%)であった。[5.1、7.1参照]
有効性評価対象となったのは、769例(ソラフェニブ群384例、プラセボ群385例)であり、組織型分類では、ソラフェニブ群377例(98.2%)、プラセボ群380例(98.7%)が淡明細胞癌であった。Motzerリスク分類では、ソラフェニブ群の200例(52.1%)、プラセボ群の194例(50.4%)が低リスク患者であり、他は中等度リスク患者であった。有効性評価対象例において、PFSの中央値はプラセボ群で84日、ソラフェニブ群で168日であった。PFSに関する解析はMotzerリスク分類及び国による層別Log-rank検定により行い、その結果、ソラフェニブのPFSに対する効果は有意であった(p<0.000001)。ハザード比(ソラフェニブ/プラセボ)は0.51(95%信頼区間:0.43〜0.60)であった。また、OSについて、イベント(死亡)数が220にて中間解析を行った結果、層別Log-rank検定のp値は0.015であり、中間解析の有意水準として設定された0.0005には至らなかったものの、ハザード比(ソラフェニブ/プラセボ)は0.71(95%信頼区間:0.54〜0.94)であり、ソラフェニブ群ではプラセボ群に比して39%の延長を示した24)。
ソラフェニブ群の安全性評価対象384例中282例(73.4%)に副作用が認められた。主な副作用は、皮疹120例(31.3%)、下痢116例(30.2%)、手足皮膚反応101例(26.3%)、脱毛87例(22.7%)であった。[5.1、7.1参照]
<切除不能な肝細胞癌>
17.1.3 海外第III相試験
全身化学療法歴のない切除又は局所療法(経皮的エタノール注入療法、ラジオ波焼灼療法、マイクロ波凝固療法、肝動脈塞栓療法/肝動脈化学塞栓療法、放射線療法等)が適用されない、Child-Pugh分類Aの肝細胞癌患者を対象として、プラセボ対照、無作為化、二重盲検により、OS等を主要評価項目、TTP(time to progression)等を副次評価項目とする試験を実施した。
有効性評価対象となったのは、602例(ソラフェニブ群299例、プラセボ群303例)であった。OSの中央値はプラセボ群241日、ソラフェニブ群324日であり、プラセボ群と比較しソラフェニブ群で有意なOSの延長が認められた(p=0.000583)。ハザード比(ソラフェニブ/プラセボ)は0.6931(95%信頼区間:0.5549〜0.8658)であった25)。
ソラフェニブ群の安全性評価対象297例中236例(79.5%)に副作用が認められた。主な副作用は、下痢116例(39.1%)、疲労64例(21.5%)、手足皮膚反応63例(21.2%)であった。[7.1参照]
有効性評価対象となったのは、602例(ソラフェニブ群299例、プラセボ群303例)であった。OSの中央値はプラセボ群241日、ソラフェニブ群324日であり、プラセボ群と比較しソラフェニブ群で有意なOSの延長が認められた(p=0.000583)。ハザード比(ソラフェニブ/プラセボ)は0.6931(95%信頼区間:0.5549〜0.8658)であった25)。
ソラフェニブ群の安全性評価対象297例中236例(79.5%)に副作用が認められた。主な副作用は、下痢116例(39.1%)、疲労64例(21.5%)、手足皮膚反応63例(21.2%)であった。[7.1参照]
<根治切除不能な甲状腺癌>
17.1.4 国際共同第III相試験
本試験への組入れ前14ヵ月以内に病勢進行が確認された局所進行又は転移性の分化型甲状腺癌(乳頭癌、濾胞癌、Hurthle細胞癌、及び低分化癌)、未分化癌又は髄様癌の所見が認められない甲状腺癌の特殊型の患者で、かつ放射性ヨウ素治療抵抗性(標的病変にヨウ素の取り込みが認められない、放射性ヨウ素治療後も標的病変における病勢進行が認められる、又は累積線量で22.2Gbq(600mCi)以上の放射性ヨウ素治療を受けている)の患者を対象として、プラセボ対照、無作為化、二重盲検により、PFSを主要評価項目、OS、TTP等を副次評価項目とする試験を実施した。なお、適切な局所治療がなされていない気管、気管支、又は食道への出血の危険性を伴う腫瘍の浸潤を認める患者は除外された。
有効性評価対象となったのは、417例(ソラフェニブ群207例、プラセボ群210例)であり、日本人患者22例(ソラフェニブ群12例、プラセボ群10例)が含まれた。PFSの中央値はプラセボ群で175日、ソラフェニブ群で329日であり、プラセボ群と比較しソラフェニブ群で有意なPFSの延長が認められた(p<0.0001)。ハザード比(ソラフェニブ/プラセボ)は0.587(95%信頼区間:0.454〜0.758)であった。
ソラフェニブ群の安全性評価対象207例中200例(96.6%)に副作用が認められた。主な副作用は、手足皮膚反応157例(75.8%)、脱毛139例(67.1%)、下痢134例(64.7%)、皮疹/落屑99例(47.8%)、疲労89例(43.0%)、高血圧77例(37.2%)、体重減少75例(36.2%)、食欲不振60例(29.0%)、粘膜炎/口内炎44例(21.3%)であった26)。[7.1参照]
有効性評価対象となったのは、417例(ソラフェニブ群207例、プラセボ群210例)であり、日本人患者22例(ソラフェニブ群12例、プラセボ群10例)が含まれた。PFSの中央値はプラセボ群で175日、ソラフェニブ群で329日であり、プラセボ群と比較しソラフェニブ群で有意なPFSの延長が認められた(p<0.0001)。ハザード比(ソラフェニブ/プラセボ)は0.587(95%信頼区間:0.454〜0.758)であった。
ソラフェニブ群の安全性評価対象207例中200例(96.6%)に副作用が認められた。主な副作用は、手足皮膚反応157例(75.8%)、脱毛139例(67.1%)、下痢134例(64.7%)、皮疹/落屑99例(47.8%)、疲労89例(43.0%)、高血圧77例(37.2%)、体重減少75例(36.2%)、食欲不振60例(29.0%)、粘膜炎/口内炎44例(21.3%)であった26)。[7.1参照]
17.1.5 国内第II相試験
根治切除不能な甲状腺未分化癌、又は局所進行若しくは転移性の甲状腺髄様癌患者を対象として、非対照、非盲検により、安全性評価を主目的とした試験を実施した。
有効性評価対象となったのは、それぞれ10例及び8例であり、治験担当医師によるRECISTに基づく奏効率は、それぞれ0%及び25.0%(95%信頼区間:3.2〜65.1%)であった。
安全性評価対象18例中18例(100%)に副作用が認められた。主な副作用は、手足皮膚反応13例(72.2%)、脱毛10例(55.6%)、高血圧9例(50.0%)、口内炎7例(38.9%)、血中甲状腺刺激ホルモン増加6例(33.3%)、発疹6例(33.3%)、斑状丘疹状皮疹6例(33.3%)、下痢5例(27.8%)、ALT増加5例(27.8%)、AST増加4例(22.2%)であった27)。[7.1参照]
有効性評価対象となったのは、それぞれ10例及び8例であり、治験担当医師によるRECISTに基づく奏効率は、それぞれ0%及び25.0%(95%信頼区間:3.2〜65.1%)であった。
安全性評価対象18例中18例(100%)に副作用が認められた。主な副作用は、手足皮膚反応13例(72.2%)、脱毛10例(55.6%)、高血圧9例(50.0%)、口内炎7例(38.9%)、血中甲状腺刺激ホルモン増加6例(33.3%)、発疹6例(33.3%)、斑状丘疹状皮疹6例(33.3%)、下痢5例(27.8%)、ALT増加5例(27.8%)、AST増加4例(22.2%)であった27)。[7.1参照]
17.2 製造販売後調査等
<切除不能な肝細胞癌>
17.2.1 日韓共同第III相試験
根治的治療不能の進行性肝細胞癌に対して肝動脈化学塞栓療法(TACE)を施行し、治療効果(腫瘍壊死効果25%以上又は腫瘍縮小率25%以上)が認められた、Child-Pugh分類Aの肝細胞癌患者を対象として、プラセボ対照、無作為化、二重盲検により、TTPを主要評価項目、OSを副次評価項目とする試験を実施した。
有効性評価対象となったのは、458例(ソラフェニブ群229例、プラセボ群229例)であり、日本人患者387例(ソラフェニブ群196例、プラセボ群191例)が含まれた。TTPの中央値はプラセボ群3.7ヵ月、ソラフェニブ群5.4ヵ月であり、プラセボ群と比較しソラフェニブ群で有意なTTPの延長は認められなかった(p=0.252)。ハザード比(ソラフェニブ/プラセボ)は0.87(95%信頼区間:0.70〜1.09)であった。
ソラフェニブ群の安全性評価対象229例中229例(100%)に副作用が認められた。主な副作用は、手足皮膚反応188例(82.1%)、リパーゼ増加101例(44.1%)、脱毛94例(41.0%)であった28)。
有効性評価対象となったのは、458例(ソラフェニブ群229例、プラセボ群229例)であり、日本人患者387例(ソラフェニブ群196例、プラセボ群191例)が含まれた。TTPの中央値はプラセボ群3.7ヵ月、ソラフェニブ群5.4ヵ月であり、プラセボ群と比較しソラフェニブ群で有意なTTPの延長は認められなかった(p=0.252)。ハザード比(ソラフェニブ/プラセボ)は0.87(95%信頼区間:0.70〜1.09)であった。
ソラフェニブ群の安全性評価対象229例中229例(100%)に副作用が認められた。主な副作用は、手足皮膚反応188例(82.1%)、リパーゼ増加101例(44.1%)、脱毛94例(41.0%)であった28)。
17.2.2 国際共同第III相試験
外科的切除術又は局所療法(ラジオ波焼灼療法又は経皮的エタノール注入療法)による根治的治療施行後に完全奏効が得られた、Child-Pugh分類A又はB(7点以下)の肝細胞癌患者を対象とした術後補助化学療法として、プラセボ対照、無作為化、二重盲検により、無再発生存期間(RFS)を主要評価項目、TTR、OSを副次評価項目とする試験を実施した。
有効性評価対象となったのは、1114例(ソラフェニブ群556例、プラセボ群558例)であり、日本人患者149例(ソラフェニブ群72例、プラセボ群77例)が含まれた。RFSの中央値はプラセボ群33.8ヵ月、ソラフェニブ群33.4ヵ月であり、プラセボ群と比較しソラフェニブ群で有意なRFSの延長は認められなかった(p=0.26)。ハザード比(ソラフェニブ/プラセボ)は0.940(95%信頼区間:0.780〜1.134)であった。
ソラフェニブ群の安全性評価対象559例中526例(94%)に副作用が認められた。主な副作用は、手足皮膚反応389例(69.6%)、下痢215例(38.5%)、脱毛179例(32.0%)であった29)。
有効性評価対象となったのは、1114例(ソラフェニブ群556例、プラセボ群558例)であり、日本人患者149例(ソラフェニブ群72例、プラセボ群77例)が含まれた。RFSの中央値はプラセボ群33.8ヵ月、ソラフェニブ群33.4ヵ月であり、プラセボ群と比較しソラフェニブ群で有意なRFSの延長は認められなかった(p=0.26)。ハザード比(ソラフェニブ/プラセボ)は0.940(95%信頼区間:0.780〜1.134)であった。
ソラフェニブ群の安全性評価対象559例中526例(94%)に副作用が認められた。主な副作用は、手足皮膚反応389例(69.6%)、下痢215例(38.5%)、脱毛179例(32.0%)であった29)。
17.3 その他
17.3.1 QT間隔に対する影響
固形癌患者31例に本剤400mgを1日2回28日間経口投与し、QT間隔延長の評価を行った。本剤の最高血中濃度到達時における、QTcF間隔のベースラインからの変化の平均値(±標準偏差)は、9.0(±18.0)msecであった。陽性対照として使用したモキシフロキサシン(400mg)の投与2時間後における、QTcF間隔のベースラインからの変化の平均値(±標準偏差)は、4.9(±13.7)msecであった。なお、QTcFが500msecを超えた例はなかった(外国人データ)。
18. 薬効薬理
18.1 作用機序
18.2 抗腫瘍効果
19. 有効成分に関する理化学的知見
19.1. ソラフェニブトシル酸塩
| 一般的名称 | ソラフェニブトシル酸塩 |
|---|---|
| 一般的名称(欧名) | Sorafenib Tosilate |
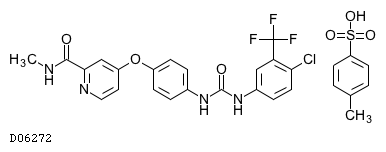
| 化学名 | 4-{4-[3-(4-Chloro-3-trifluoromethylphenyl)ureido]phenoxy}-N2-methylpyridine-2-carboxamide mono(4-methylbenzenesulfonate) |
| 分子式 | C21H16ClF3N4O3・C7H8O3S |
| 分子量 | 637.03 |
| 融点 | 223〜231℃ |
| 物理化学的性状 | 本品は白色〜帯黄白色又は帯褐白色の粉末である。 本品はジメチルスルホキシド又はN,N-ジメチルホルムアミドに溶けやすく、メタノール又はエタノール(99.5)に溶けにくく、水にほとんど溶けない。 |
| 分配係数 | 本品はオクタノール/水及びオクタノール/リン酸塩緩衝液にほとんど溶けないため、分配係数を測定することはできなかった。 |
| KEGG DRUG |
|
22. 包装
60錠[10錠(PTP)×6]
23. 主要文献
- 社内資料:ラットにおける生殖発生毒性試験(2008年1月25日承認、CTD2.6.6.6.1)
- 社内資料:ウサギにおける生殖発生毒性試験(2008年1月25日承認、CTD2.6.6.6.2)
- 社内資料:イヌにおける反復投与毒性試験(2008年1月25日承認、CTD2.6.6.3.2.1)
- Mross K,et al., Eur J Cancer., 43, 55-63, (2007) »PubMed »DOI
- 社内資料:ドキソルビシンとの相互作用(2008年1月25日承認、CTD2.7.2.2.4.3.3)
- 社内資料:リファンピシンとの相互作用
- 社内資料:ワルファリンとの相互作用(2008年1月25日承認、CTD2.7.2.3.4.2.2)
- 社内資料:ドセタキセルとの相互作用
- 社内資料:フラジオマイシン(ネオマイシン)との相互作用(2014年6月20日承認、CTD2.7.6.14)
- NDBを用いた調査結果の概要(VEGF/VEGFR阻害作用を有する薬剤の動脈解離に関するリスク評価), (https://www.pmda.go.jp/files/000266521.pdf)
- 社内資料:ラットにおける反復投与毒性試験(2008年1月25日承認、CTD2.6.6.3.1.1)
- 社内資料:ラットにおける反復投与毒性試験(2008年1月25日承認、CTD2.6.6.3.1.2)
- 社内資料:イヌにおける反復投与毒性試験(2008年1月25日承認、CTD2.6.6.3.2.3)
- 社内資料:薬物動態(2008年1月25日承認、CTD2.7.2.2.1)
- 社内資料:食事の影響(外国人)(2008年1月25日承認、CTD2.7.1.2.3)
- 社内資料:タンパク結合(2008年1月25日承認、CTD2.6.4.4.1)
- 社内資料:薬物動態(外国人)(2008年1月25日承認、CTD2.7.2.2.3.1)
- 社内資料:腎機能障害のある被験者における薬物動態(外国人)
- Furuse J,et al., Cancer Sci., 99, 159-165, (2008) »PubMed
- 社内資料:薬物間相互作用(in vitro)(2008年1月25日承認、CTD2.6.4.5.1.5)
- Lathia C,et al., Cancer Chemother Pharmacol., 57, 685-692, (2006) »PubMed
- 社内資料:薬物間相互作用(外国人)(2008年1月25日承認、CTD2.7.2.2.4.2)
- 社内資料:腎細胞癌患者を対象とした第II相臨床試験(2008年1月25日承認、CTD2.7.6.23)
- Escudier B,et al., N Engl J Med., 356, 125-134, (2007) »PubMed
- Llovet JM,et al., N Engl J Med., 359, 378-390, (2008) »PubMed
- 社内資料:甲状腺癌患者を対象とした第III相臨床試験(2014年6月20日承認、CTD2.7.6.1)
- 社内資料:甲状腺未分化癌及び甲状腺髄様癌患者を対象とした第II相臨床試験
- Kudo M,et al., Eur J Cancer., 47, 2117-2127, (2011) »PubMed
- 社内資料:肝細胞癌患者を対象とした市販後の国際共同第III相臨床試験
- Wilhelm SM,et al., Cancer Research., 64, 7099-7109, (2004) »PubMed
- Chang YS,et al., Cancer Chemother Pharmacol., 59, 561-574, (2007) »PubMed
- Liu L,et al., Cancer Research., 66, 11851-11858, (2006) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
バイエル薬品株式会社
メディカルインフォメーション
〒530-0001
大阪市北区梅田二丁目4番9号
文献請求先
製品情報問い合わせ先
バイエル薬品株式会社
電話:0120-106-398
バイエル医療用医薬品のお問い合わせ先
26. 製造販売業者等
26.1 製造販売元
バイエル薬品株式会社
大阪市北区梅田二丁目4番9号