医薬品情報
| 総称名 | サイバインコ |
|---|---|
| 一般名 | アブロシチニブ |
| 欧文一般名 | Abrocitinib |
| 製剤名 | アブロシチニブ錠 |
| 薬効分類名 | ヤヌスキナーゼ(JAK)阻害剤 |
| 薬効分類番号 | 4490 |
| ATCコード | D11AH08 |
| KEGG DRUG |
D11400
アブロシチニブ
|
| KEGG DGROUP |
DG02020
JAK阻害薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2023年7月 改訂(第4版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| サイバインコ錠50mg | CIBINQO Tablets | ファイザー | 4490037F1026 | 2180.6円/錠 | 劇薬, 処方箋医薬品注) |
| サイバインコ錠100mg | CIBINQO Tablets | ファイザー | 4490037F2022 | 4245.6円/錠 | 劇薬, 処方箋医薬品注) |
| サイバインコ錠200mg | CIBINQO Tablets | ファイザー | 4490037F3029 | 6384.5円/錠 | 劇薬, 処方箋医薬品注) |
1. 警告
1.1 本剤投与により、結核、肺炎、敗血症、ウイルス感染等による重篤な感染症の新たな発現もしくは悪化等が報告されており、本剤との関連性は明らかではないが、悪性腫瘍の発現も報告されている。本剤が疾病を完治させる薬剤でないことも含め、これらの情報を患者に十分説明し、患者が理解したことを確認した上で、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。
また、本剤投与により重篤な副作用が発現し、致命的な経過をたどることがあるので、緊急時の対応が十分可能な医療施設及び医師が使用し、本剤投与後に副作用が発現した場合には、主治医に連絡するよう患者に注意を与えること。[2.2、2.3、8.1-8.4、8.7、9.1.1、9.1.2、9.1.5、9.8、11.1.1、15.1.1、15.1.2参照]
また、本剤投与により重篤な副作用が発現し、致命的な経過をたどることがあるので、緊急時の対応が十分可能な医療施設及び医師が使用し、本剤投与後に副作用が発現した場合には、主治医に連絡するよう患者に注意を与えること。[2.2、2.3、8.1-8.4、8.7、9.1.1、9.1.2、9.1.5、9.8、11.1.1、15.1.1、15.1.2参照]
1.2 感染症
1.2.1 重篤な感染症
1.2.2 結核
ヤヌスキナーゼ(JAK)阻害剤において、播種性結核(粟粒結核)及び肺外結核(脊椎、リンパ節等)を含む結核が報告されている。結核の既感染者では症状の顕在化及び悪化のおそれがあるため、本剤投与に先立って結核に関する十分な問診及び胸部X線検査に加え、インターフェロン-γ遊離試験又はツベルクリン反応検査を行い、適宜胸部CT検査等を行うことにより、結核感染の有無を確認すること。結核の既往歴を有する患者及び結核の感染が疑われる患者には、結核等の感染症について診療経験を有する医師と連携の下、原則として本剤の投与開始前に適切な抗結核薬を投与すること。
ツベルクリン反応等の検査が陰性の患者に投与後活動性結核が認められた例も報告されている。[2.3、8.3、9.1.2、11.1.1参照]
ツベルクリン反応等の検査が陰性の患者に投与後活動性結核が認められた例も報告されている。[2.3、8.3、9.1.2、11.1.1参照]
1.3 本剤についての十分な知識と適応疾患の治療の知識・経験を持つ医師が使用すること。
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.9 妊婦又は妊娠している可能性のある女性[9.5参照]
4. 効能または効果
既存治療で効果不十分なアトピー性皮膚炎
5. 効能または効果に関連する注意
5.1 ステロイド外用剤やタクロリムス外用剤等の抗炎症外用剤による適切な治療を一定期間施行しても、十分な効果が得られず、強い炎症を伴う皮疹が広範囲に及ぶ患者に用いること。
5.2 原則として、本剤投与時にはアトピー性皮膚炎の病変部位の状態に応じて抗炎症外用剤を併用すること。
5.3 本剤投与時も保湿外用剤を継続使用すること。[8.11参照]
6. 用法及び用量
通常、成人及び12歳以上の小児には、アブロシチニブとして100mgを1日1回経口投与する。なお、患者の状態に応じて200mgを1日1回投与することができる。
7. 用法及び用量に関連する注意
7.1 中等度の腎機能障害(30≦eGFR〔推算糸球体ろ過量:mL/分/1.73m2〕<60)及び重度の腎機能障害(eGFR<30)を有する患者には、50mgを1日1回経口投与すること。中等度の腎機能障害を有する患者においては、患者の状態に応じて100mgを1日1回投与することができる。[9.2.1、9.2.2、16.6.1参照]
7.3 本剤による治療反応は、通常投与開始から12週までには得られる。12週までに治療反応が得られない場合は、投与中止を考慮すること。
7.4 免疫抑制作用が増強されると感染症のリスクが増加することが予想されるので、本剤と適応疾患の生物製剤、他の経口JAK阻害剤、シクロスポリン等の免疫抑制剤(局所製剤以外)との併用はしないこと。本剤とこれらの薬剤との併用経験はない。
8. 重要な基本的注意
8.1 本剤は、免疫反応に関与するJAKファミリーを阻害するので、感染症に対する宿主免疫能に影響を及ぼす可能性がある。本剤の投与に際しては十分な観察を行い、感染症の発現や増悪に注意すること。また、患者に対し、発熱、倦怠感等があらわれた場合には、速やかに主治医に相談するよう指導すること。[1.1、1.2.1、2.2、9.1.1、9.1.5、11.1.1参照]
8.2 本剤は免疫抑制作用を有することから、皮膚バリア機能が低下しているアトピー性皮膚炎患者への投与に際しては十分な観察を行い、皮膚感染症の発現に注意すること。アトピー性皮膚炎患者を対象とした臨床試験において重篤な皮膚感染症が報告されている。[1.1、1.2.1、9.8、11.1.1参照]
8.3 本剤投与に先立って結核に関する十分な問診及び胸部X線検査に加え、インターフェロン-γ遊離試験又はツベルクリン反応検査を行い、適宜胸部CT検査等を行うことにより、結核感染の有無を確認すること。本剤投与中は胸部X線検査等の適切な検査を定期的に行うなど結核の発現には十分に注意し、患者に対し、結核を疑う症状が発現した場合(持続する咳、発熱等)には速やかに主治医に連絡するよう説明すること。[1.1、1.2.2、2.3、9.1.2、11.1.1参照]
8.4 ヘルペスウイルスを含むウイルスの再活性化(帯状疱疹、単純ヘルペス等)が報告されている。また、重篤な帯状疱疹や播種性帯状疱疹も認められていることから、ヘルペスウイルス等の再活性化の徴候や症状の発現に注意すること。徴候や症状の発現が認められた場合には、患者に受診するよう説明し、本剤の投与を中断し速やかに適切な処置を行うこと。また、ヘルペスウイルス以外のウイルスの再活性化にも注意すること。[1.1、1.2.1、9.8、11.1.1参照]
8.5 JAK阻害剤によるB型肝炎ウイルスの再活性化が報告されているので、本剤投与に先立って、B型肝炎ウイルス感染の有無を確認すること。[9.1.3参照]
8.6 感染症発現のリスクを否定できないので、本剤開始直前及び投与中の生ワクチンの接種は行わないこと。
8.8 好中球減少、リンパ球減少、ヘモグロビン減少及び血小板減少があらわれることがあるので、本剤の投与開始前及び投与開始後は定期的に好中球数、リンパ球数、血小板数及びヘモグロビン値を確認すること。[2.5-2.8、9.1.7-9.1.10、9.8、11.1.3参照]
8.9 総コレステロール、LDLコレステロール、HDLコレステロール及びトリグリセリドの上昇等の脂質検査値異常があらわれることがある。本剤投与開始後は定期的に脂質検査値を確認すること。臨床上必要と認められた場合には、高脂血症治療薬の投与等の適切な処置を考慮すること。
8.10 肝機能障害があらわれることがあるので、トランスアミナーゼ値上昇に注意するなど観察を十分に行うこと。[11.1.5参照]
8.11 本剤が疾病を完治させる薬剤でなく、本剤投与中も保湿外用剤等を併用する必要があることを患者に対して説明し、患者が理解したことを確認したうえで投与すること。[5.3参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
(1)結核の既感染者では、結核を活動化させるおそれがある。
(2)結核の既往歴を有する場合及び結核感染が疑われる場合には、結核の診療経験がある医師に相談すること。以下のいずれかの患者には、原則として本剤の開始前に適切な抗結核薬を投与すること。
・胸部画像検査で陳旧性結核に合致するか推定される陰影を有する患者
・結核の治療歴(肺外結核を含む)を有する患者
・インターフェロン-γ遊離試験やツベルクリン反応検査等の検査により、既感染が強く疑われる患者
・結核患者との濃厚接触歴を有する患者
9.1.3 B型肝炎ウイルスキャリアの患者又は既往感染者(HBs抗原陰性、かつHBc抗体又はHBs抗体陽性)
肝機能検査値やHBV DNAのモニタリングを行うなど、B型肝炎ウイルスの再活性化の徴候や症状の発現に注意すること。JAK阻害剤を投与されたB型肝炎ウイルスキャリアの患者又は既往感染者において、B型肝炎ウイルスの再活性化が報告されている。[8.5参照]
9.1.4 C型肝炎患者
HCV抗体陽性、HCV RNA陽性の患者は臨床試験から除外されている。
9.1.5 易感染性の状態にある患者
9.1.6 静脈血栓塞栓症のリスクを有する患者
深部静脈血栓症及び肺塞栓症が報告されている。[11.1.2参照]
9.1.7 好中球減少(好中球数1,000/mm3未満を除く)のある患者
9.1.8 リンパ球減少(リンパ球数500/mm3未満を除く)のある患者
9.1.9 ヘモグロビン値減少(ヘモグロビン値8g/dL未満を除く)のある患者
9.1.10 血小板減少(血小板数50,000/mm3未満の患者を除く)のある患者
9.1.11 間質性肺炎の既往歴のある患者
定期的に問診を行うなど、注意すること。間質性肺炎があらわれるおそれがある。[11.1.4参照]
9.1.12 腸管憩室のある患者
消化管穿孔があらわれるおそれがある。[11.1.6参照]
9.2 腎機能障害患者
9.2.1 中等度の腎機能障害(30≦eGFR〔mL/分/1.73m2〕<60)を有する患者
9.2.2 重度の腎機能障害(eGFR<30)を有する患者
9.3 肝機能障害患者
9.4 生殖能を有する者
妊娠可能な女性は、本剤投与中及び本剤投与終了後一定期間は適切な避妊を行うよう指導すること。ラットを用いた受胎能試験において、妊娠率の低下、黄体数及び着床数の減少、着床後胚損失率の上昇を含めた受胎能への影響が認められ、このときの血漿中薬物濃度はアトピー性皮膚炎患者に本剤200mgを1日1回投与したときの血漿中濃度と比較したとき7倍程度であった1)。
9.5 妊婦
9.6 授乳婦
9.7 小児等
12歳未満の小児等を対象とした臨床試験は実施していない。
9.8 高齢者
10. 相互作用
相互作用序文
本剤は主にCYP2C19及びCYP2C9で代謝される。また、本剤はCYP2C19に対して阻害作用を示す。[16.4、16.7.2参照]
薬物代謝酵素用語
CYP2C19
薬物代謝酵素用語
CYP2C9
10.2 併用注意
| CYP2C19の強い阻害薬 フルコナゾール、フルボキサミン、チクロピジン [7.2、16.7.1参照] | 本剤の作用が増強する可能性があるので、これらの薬剤は可能な限り他の類薬に変更する、又はこれらの薬剤を休薬する等を考慮すること。 | これらの薬剤がCYP2C19の代謝活性を阻害するため、アブロシチニブの血中濃度が上昇する可能性がある。 |
| CYP2C19及びCYP2C9の強い又は中程度の誘導薬 リファンピシン等 [16.7.1参照] | 本剤の効果が減弱する可能性があるので、これらの薬剤は誘導作用のない又は弱い他の類薬に変更する等を考慮すること。 | これらの薬剤がCYP2C19及びCYP2C9の代謝活性を誘導するため、アブロシチニブの血中濃度が低下する可能性がある。 |
| P-gpの基質となる薬剤 ダビガトランエテキシラート、ジゴキシン等 [16.7.2参照] | これらの薬剤の作用が増強されるおそれがあるので、患者の状態を慎重に観察し、副作用発現に十分注意すること。 | 本剤がP-gpを阻害することにより、これらの薬剤の血漿中濃度が上昇する可能性がある。 |
| クロピドグレル | クロピドグレルの作用が減弱されるおそれがあるので、併用する際には注意すること。 | 本剤がCYP2C19を阻害することにより、クロピドグレルの活性代謝物の血中濃度が低下する。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 感染症
単純ヘルペス(3.2%)注1)、帯状疱疹(1.6%)注2)、肺炎(0.2%)、結核(頻度不明)等の重篤な感染症があらわれ、致死的な経過をたどることがある。重篤な感染症、敗血症、日和見感染を発現した場合には、感染症がコントロールできるようになるまで本剤を休薬すること。[1.1、1.2.1、1.2.2、2.2、2.3、8.1-8.4、9.1.1、9.1.2、9.1.5、9.8、15.1.1参照]
注1)口腔ヘルペス、単純ヘルペス、眼部単純ヘルペス、ヘルペス眼感染、ヘルペス性皮膚炎、鼻ヘルペスを含む
注2)帯状疱疹、眼帯状疱疹を含む
11.1.2 静脈血栓塞栓症
肺塞栓症(0.1%未満)及び深部静脈血栓症(0.1%未満)を含む静脈血栓塞栓症があらわれることがある。[9.1.6参照]
11.1.3 血小板減少(1.4%)、ヘモグロビン減少(ヘモグロビン減少0.9%、貧血0.6%)、リンパ球減少(0.7%)、好中球減少(0.4%)
血小板数
本剤投与開始後、50,000/mm3未満になった場合には、投与を中止すること。
ヘモグロビン値
本剤投与開始後、8g/dL未満になった場合には、8g/dL以上に回復するまで休薬すること。
リンパ球数
本剤投与開始後、500/mm3未満になった場合には、500/mm3以上に回復するまで休薬すること。
好中球数
本剤投与開始後、1,000/mm3未満になった場合には、1,000/mm3以上に回復するまで休薬すること。
11.1.4 間質性肺炎(0.1%)
発熱、咳嗽、呼吸困難等の呼吸器症状に十分に注意し、異常が認められた場合には、速やかに胸部X線検査、胸部CT検査及び血液ガス検査等を実施し、本剤の投与を中止するとともにニューモシスチス肺炎との鑑別診断(β-Dグルカンの測定等)を考慮に入れ適切な処置を行うこと。[9.1.11参照]
11.1.5 肝機能障害
ALT(0.8%)、AST(0.6%)の上昇等を伴う肝機能障害(頻度不明)があらわれることがある。[8.10参照]
11.1.6 消化管穿孔(頻度不明)
異常が認められた場合には投与を中止するとともに、腹部X線、CT等の検査を実施するなど十分に観察し、適切な処置を行うこと。[9.1.12参照]
11.2 その他の副作用
| 1%以上 | 1%未満 | |
| 胃腸障害 | 悪心(11.0%)、腹痛、嘔吐、下痢 | 消化不良、腹部不快感、胃食道逆流性疾患、腹部膨満 |
| 一般・全身障害及び投与部位の状態 | 疲労 | 無力症、発熱 |
| 感染症及び寄生虫症 | 上咽頭炎、上気道感染、毛包炎 | 尿路感染、結膜炎、ヘルペス性状湿疹、膿痂疹、インフルエンザ、咽頭炎、副鼻腔炎、気管支炎、膀胱炎、せつ、膿瘍、皮膚感染、胃腸炎、下気道感染、感染性湿疹、皮膚真菌感染 |
| 血液及びリンパ系障害 | 白血球減少、リンパ節症、赤血球減少、白血球増加 | |
| 血管障害 | 高血圧 | |
| 呼吸器、胸郭及び縦隔障害 | 咳嗽、鼻出血 | |
| 心臓障害 | 動悸、心室内伝導障害 | |
| 神経系障害 | 頭痛(4.4%)、浮動性めまい | 傾眠 |
| 代謝及び栄養障害 | 体重増加、高脂血症(脂質異常症を含む) | |
| 皮膚及び皮下組織障害 | ざ瘡(3.6%) | 脱毛症、蕁麻疹、そう痒症 |
| 良性、悪性及び詳細不明の新生物(嚢胞及びポリープを含む) | 皮膚乳頭腫(疣贅等) | |
| 臨床検査 | 血中CK増加 | NK細胞減少、LDH増加、γ-GT上昇、尿中蛋白陽性、プロトロンビン時間延長 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.1 臨床使用に基づく情報
15.1.1 アトピー性皮膚炎患者を対象とした後期第2相試験1試験及び第3相試験6試験の併合解析において、重篤な感染症の発現頻度(因果関係問わない)は、本剤100mg投与群で1.9%(19/1023例)、本剤200mg投与群で1.3%(27/2105例)であり、100人年あたりの発現率(95%信頼区間)は本剤100mg投与群で2.18(1.31,3.40)、本剤200mg投与群で2.11(1.39,3.07)であった。また、悪性腫瘍(非黒色腫皮膚癌を除く)の発現頻度(因果関係問わない)は、本剤100mg投与群で0.1%(1/1023例)、本剤200mg投与群で0.1%(2/2105例)であり、100人年あたりの発現率(95%信頼区間)は本剤100mg投与群で0.11(0.00,0.63)、本剤200mg投与群で0.16(0.02,0.56)であった。[1.1、1.2.1、2.2、8.7、11.1.1参照]
15.2 非臨床試験に基づく情報
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与時
16.1.2 反復投与時
母集団薬物動態モデルを用いたシミュレーションの結果、日本人アトピー性皮膚炎患者に本剤100mg及び200mgを1日1回反復投与したときのアブロシチニブのAUCtauはそれぞれ3680及び8280ng・h/mL、Cmaxはそれぞれ740及び1580ng/mLであった7)。
16.2 吸収
16.2.1 バイオアベイラビリティ
健康成人6例に本剤200mgを単回経口投与及び80μgを単回静脈内投与したときのアブロシチニブの絶対的バイオアベイラビリティは約60%(90%信頼区間:46%〜78%)であった8)(外国人データ)。
16.2.2 食事の影響
健康成人15例に本剤200mgを食後(高脂肪食)投与したとき、空腹時投与と比較して、アブロシチニブのAUCinf及びCmaxはそれぞれ約26%及び29%増加した9)(外国人データ)。
16.3 分布
16.4 代謝
16.5 排泄
16.6 特定の背景を有する患者
16.6.1 腎機能障害
中等度(30≦eGFR〔mL/分〕<60:7例)の腎機能障害を有する被験者に本剤200mgを単回経口投与したとき、腎機能正常被験者(eGFR≧90:8例)と比較して、アブロシチニブ、活性代謝物のM1及びM2、ならびにこれらの活性成分の非結合型曝露量(それぞれの相対力価で補正)の総和(以下、活性成分)のAUCinfはそれぞれ約83%、54%、170%及び110%増加した。重度(eGFR<30:8例)の腎機能障害を有する被験者に本剤200mgを単回経口投与したとき、腎機能正常被験者と比較して、アブロシチニブ、M1及びM2、ならびに活性成分のAUCinfはそれぞれ約21%、187%、471%及び191%増加した(外国人データ)。これらの結果から、軽度(60≦eGFR<90)の腎機能障害を有する被験者のeGFRが60の場合、アブロシチニブ、M1及びM2、ならびに活性成分のAUCinfは約29%、61%、138%及び70%増加すると推定された13)。[7.1、9.2.1、9.2.2参照]
16.6.2 肝機能障害
16.7 薬物相互作用
16.7.1 併用薬がアブロシチニブの薬物動態に及ぼす影響
アブロシチニブ、M1及びM2、ならびに活性成分の曝露量に及ぼす併用薬の影響を下表に示す(外国人データ)。[7.2、10.2参照]
(1)フルコナゾール(CYP2C19の強い阻害薬、CYP2C9及びCYP3Aの中程度の阻害薬)5)
| 併用薬投与量 | 400mg(1日目) 200mg(2〜7日目) | |
| 本剤投与量 | 100mg単回 | |
| 例数 | 12 | |
| 薬物動態パラメータ調整済み幾何平均値の比(%)[90%信頼区間]併用/単独 | ||
| 活性成分a) | Cmax | 123.46[107.58,141.70] |
| AUCinf | 254.86[241.75,268.67] | |
| アブロシチニブ | Cmax | 192.10[154.15,239.39] |
| AUCinf | 482.86[383.94,607.26] | |
| M1 | Cmax | 9.50[7.81,11.55] |
| AUCinf | 25.87[22.87,29.27] | |
| M2 | Cmax | 23.83[19.97,28.42] |
| AUCinf | 61.19[37.56,99.69] | |
(2)フルボキサミン(CYP2C19の強い阻害薬、CYP3Aの中程度の阻害薬)5)
| 併用薬投与量 | 50mg 1日1回9日間 | |
| 本剤投与量 | 100mg単回 | |
| 例数 | 12 | |
| 薬物動態パラメータ調整済み幾何平均値の比(%)[90%信頼区間]併用/単独 | ||
| 活性成分a) | Cmax | 133.08[99.58,177.86] |
| AUCinf | 191.24[173.81,210.43] | |
| アブロシチニブ | Cmax | 184.44[133.27,255.24] |
| AUCinf | 275.22[238.77,317.24] | |
| M1 | Cmax | 41.62[32.30,53.63] |
| AUCinf | 78.96[72.75,85.70] | |
| M2 | Cmax | 71.30[58.60,86.75] |
| AUCinf | 112.79[105.59,120.49] | |
(3)リファンピシン(CYP2C19、CYP2C9及びCYP3A4の強い誘導薬)15)
| 併用薬投与量 | 600mg 1日1回8日間 | |
| 本剤投与量 | 200mg単回 | |
| 例数 | 12 | |
| 薬物動態パラメータ調整済み幾何平均値の比(%)[90%信頼区間]併用/単独 | ||
| 活性成分a) | Cmax | 68.91[50.28,94.46] |
| AUCinf | 43.86[40.94,46.98] | |
| アブロシチニブ | Cmax | 20.86[14.31,30.41] |
| AUCinf | 12.45[9.33,16.60] | |
| M1 | Cmax | 168.36[115.54,245.32] |
| AUCinf | 94.80[80.11,112.19] | |
| M2 | Cmax | 145.45[102.97,205.45] |
| AUCinf | 72.95[68.39,77.83] | |
(4)プロベネシド(OAT3の阻害薬)6)
| 併用薬投与量 | 1000mg 1日2回3日間 | |
| 本剤投与量 | 200mg単回 | |
| 例数 | 12 | |
| 薬物動態パラメータ調整済み幾何平均値の比(%)[90%信頼区間]併用/単独 | ||
| 活性成分a) | Cmax | 130.13[104.10,162.65] |
| AUCinf | 165.54[152.00,180.29] | |
| アブロシチニブ | Cmax | 121.38[92.93,158.52] |
| AUCinf | 127.60[114.97,141.61] | |
| M1 | Cmax | 136.69[116.33,160.61] |
| AUCinf | 177.17[164.48,190.84] | |
| M2 | Cmax | 134.60[115.08,157.44] |
| AUCinf | 224.85[207.95,243.12] | |
16.7.2 アブロシチニブが併用薬の薬物動態に及ぼす影響
In vitro試験において、アブロシチニブはCYP3A、CYP2C19及びCYP2D6に対して弱い時間依存的阻害作用16)を示し、CYP3A4に対して弱い誘導作用を示した17)。アブロシチニブはOATP1B1/1B3、OAT1、OCT2及びBSEPを阻害しなかったが、OAT3、P-gp、BCRP、OCT1、MATE1及びMATE2Kを阻害した18)。薬物相互作用を検討した臨床試験の結果、アブロシチニブはP-gp及びCYP2C19を阻害した。
アブロシチニブが併用薬の薬物動態に及ぼす影響を下表に示す(外国人データ)。[10.、10.2参照]
アブロシチニブが併用薬の薬物動態に及ぼす影響を下表に示す(外国人データ)。[10.、10.2参照]
| 併用薬 | 併用薬投与量 | 本剤投与量 | 例数 | 併用薬の薬物動態パラメータ 調整済み幾何平均値の比(%)[90%信頼区間]併用/単独 | |
| Cmaxa) | AUCinf | ||||
| エチニルエストラジオール (経口避妊薬)19) | 30μg単回 | 200mg 1日1回11日間 | 15 | 107.17[99.17,115.82] | 118.78[111.98,125.99] |
| レボノルゲストレル (経口避妊薬)19) | 150μg単回 | 200mg 1日1回11日間 | 15 | 86.02[75.75,97.67] | 97.57b)[86.56,109.99] |
| ミダゾラム (CYP3A4及びCYP3A5の基質)20) | 2mg単回 (2日目投与) | 200mg 1日1回7日間 | 25、24c) | 86.29[77.27,96.36] | 84.28[78.95,89.97] |
| 2mg単回 (7日目投与) | 93.54[83.76,104.46] | 92.29[86.45,98.52] | |||
| ダビガトランエテキシラート (P-gpの基質)21) | 75mg単回 | 200mg 単回 | 20 | 140.10[92.20,212.90] | 152.86[108.79,214.80] |
| ロスバスタチン (BCRP及びOAT3の基質)22) | 10mg単回 | 200mg 1日1回3日間 | 12 | 91.27[82.67,100.77] | 101.94[92.89,111.88] |
| メトホルミン (OCTs、MATE1及びMATE2Kの基質)23) | 500mg単回 | 200mg 1日1回2日間 | 12 | 98.50[82.09,118.20] | 94.25b)[88.19,100.73] |
| カフェインd) (CYP1A2の基質)24) | 100mg単回 | 200mg 1日1回10日間 | 13 | 101.22[92.21,111.12] | 139.59e)[121.98,159.74] |
| エファビレンツd) (CYP2B6の基質)24) | 50mg単回 | 200mg 1日1回10日間 | 13 | 97.26[83.25,113.62] | 110.10b)[103.45,117.17] |
| オメプラゾール (CYP2C19の基質)24) | 10mg単回 | 200mg 1日1回10日間 | 13 | 234.16[170.19,322.17] | 288.81[240.56,346.73] |
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第III相試験単剤投与試験
ステロイド外用剤又はタクロリムス外用剤等の外用剤治療で効果不十分、又は外用薬治療が医学的に不適切、あるいは疾患コントロールのために全身療法を必要とする12歳以上の中等症から重症注1)のアトピー性皮膚炎(AD)患者391例(日本人患者44例を含む)を対象に、本剤200mg、本剤100mg又はプラセボを1日1回、12週間投与した注2)。主要評価項目とした投与後12週時点のIGA改善達成注3)及びEASI-75達成注4)において、本剤の両用量群はプラセボ群に比べて統計的に有意な改善効果を示した。
注1)IGAスコアが3以上、EASIスコアが16以上、及び体表面積に占めるAD病変の割合が10%以上、そう痒の重症度のNRSスコアが4以上
注2)投与期間中は保湿剤の併用は許容されており、経口シクロスポリン、経口ステロイド等の全身療法及び光線療法の併用を禁止した
注3)IGAスコアが「消失」(スコア0)又は「ほぼ消失」(スコア1)と判定され、かつベースライン時から2段階以上の改善達成
注4)EASIスコアのベースライン時からの75%以上の改善達成
| 本剤100mg群 | 本剤200mg群 | プラセボ群 | ||
| IGA改善達成 | 12週時点のIGA改善達成率a) | 28.4(44/155) | 38.1(59/155) | 9.1(7/77) |
| プラセボ群との差[95%信頼区間]b) 調整p値c),d) | 19.3[9.6,29.0] 0.0008 | 28.7[18.6,38.8] <0.0001 | − | |
| EASI-75達成 | 12週時点のEASI-75達成率a) | 44.5(69/155) | 61.0(94/154) | 10.4(8/77) |
| プラセボ群との差[95%信頼区間]b) 調整p値c),d) | 33.9[23.3,44.4] <0.0001 | 50.5[40.0,60.9] <0.0001 | − | |
| PP-NRS4達成e) | 12週時点のPP-NRS4達成率f) | 45.2 | 55.3 | 11.5 |
| プラセボ群との差[95%信頼区間]b) | 33.7[22.8,44.7] | 43.9[32.9,55.0] | − |
副作用発現頻度は、本剤200mg投与群で34.8%(54/155例)及び本剤100mg投与群では19.6%(31/158例)であった。主な副作用は本剤200mg投与群では悪心13.5%(21/155例)及び頭痛5.8%(9/155例)、本剤100mg投与群では悪心4.4%(7/158例)及び頭痛2.5%(4/158例)であった25)。
17.1.2 国際共同第III相試験外用剤併用投与試験(成人)
ステロイド外用剤又はタクロリムス外用剤等の外用剤治療で効果不十分、あるいは疾患コントロールのために全身療法を必要とする18歳以上の中等症から重症注1)のAD患者837例(日本人患者76例を含む)を対象に、ステロイド外用剤併用下で、本剤200mg、本剤100mgを1日1回とデュピルマブに対応するプラセボを隔週投与、デュピルマブ300mgの隔週投与(初回用量は600mg)と本剤に対応するプラセボを1日1回、又は第1日から16週間本剤に対応するプラセボを1日1回とデュピルマブに対応するプラセボを隔週投与で16週間投与した注2)。主要評価項目とした投与後12週時点のIGA改善達成注3)及びEASI-75達成注4)において、本剤の両用量群はプラセボ群に比べて統計的に有意な改善効果を示した。
注1)IGAスコアが3以上、EASIスコアが16以上、及び体表面積に占めるAD病変の割合が10%以上、そう痒の重症度のNRSスコアが4以上
注2)投与期間中は保湿剤の併用を必須とし、経口シクロスポリン、経口ステロイド等の全身療法及び光線療法の併用を禁止した
注3)IGAスコアが「消失」(スコア0)又は「ほぼ消失」(スコア1)と判定され、かつベースライン時から2段階以上のIGAスコアの改善達成
注4)EASIスコアのベースライン時からの75%以上の改善達成
| 本剤100mg群 | 本剤200mg群 | デュピルマブ群 | プラセボ群 | ||
| IGA改善達成 | 12週時点のIGA改善達成率a) | 36.6(86/235) | 48.4(106/219) | 36.5(88/241) | 14.0(18/129) |
| プラセボ群との差[95%信頼区間]b) 調整p値c),d) | 23.1[14.7,31.4] <0.0001 | 34.8[26.1,43.5] <0.0001 | − | − | |
| EASI-75達成 | 12週時点のEASI-75達成率a) | 58.7(138/235) | 70.3(154/219) | 58.1(140/241) | 27.1(35/129) |
| プラセボ群との差[95%信頼区間]b) 調整p値c),d) | 31.9[22.2,41.6] <0.0001 | 43.2[33.7,52.7] <0.0001 | − | − | |
| PP-NRS4達成e) | 12週時点のPP-NRS4達成率a) | 47.5(105/221) | 63.1(137/217) | 54.5(122/224) | 28.9(35/121) |
| プラセボ群との差[95%信頼区間]b) | 18.5[8.0,28.9] | 33.7[23.4,44.1] | − | − |
副作用発現頻度は、本剤200mg投与群で29.6%(67/226例)及び本剤100mg投与群では19.7%(47/238例)であった。主な副作用は本剤200mg投与群では悪心10.2%(23/226例)、頭痛3.5%(8/226例)及びざ瘡3.5%(8/226例)、本剤100mg投与群では上咽頭炎3.8%(9/238例)及び悪心2.1%(5/238例)であった26)。
17.1.3 国際共同第III相試験外用剤併用投与試験(青少年)
ステロイド外用剤又はタクロリムス外用剤等の外用剤治療で効果不十分、あるいは疾患コントロールのために全身療法を必要とする12歳以上18歳未満の中等症から重症注1)のAD患者285例(日本人患者26例を含む)を対象に、ステロイド外用剤併用下で、本剤200mg、本剤100mg又はプラセボを1日1回、12週間投与した注2)。主要評価項目とした投与後12週時点のIGA改善達成注3)及びEASI-75達成注4)において、本剤の両用量群はプラセボ群に比べて統計的に有意な改善効果を示した。
注1)IGAスコアが3以上、EASIスコアが16以上、及び体表面積に占めるAD病変の割合が10%以上、そう痒の重症度のNRSスコアが4以上
注2)投与期間中は保湿剤の併用を必須とし、経口シクロスポリン、経口ステロイド等の全身療法及び光線療法の併用を禁止した
注3)IGAスコアが「消失」(スコア0)又は「ほぼ消失」(スコア1)と判定され、かつベースライン時から2段階以上のIGAスコアの改善達成
注4)EASIスコアのベースライン時からの75%以上の改善達成
| 本剤100mg群 | 本剤200mg群 | プラセボ群 | ||
| IGA改善達成 | 12週時点のIGA改善達成率a) | 41.6(37/89) | 46.2(43/93) | 24.5(23/94) |
| プラセボ群との差[95%信頼区間]b) 調整p値c),d) | 16.7[3.5,29.9] 0.0147 | 20.6[7.3,33.9] 0.0030 | − | |
| EASI-75達成 | 12週時点のEASI-75達成率a) | 68.5(61/89) | 72.0(67/93) | 41.5(39/94) |
| プラセボ群との差[95%信頼区間]b) 調整p値c),d) | 26.5[13.1,39.8] 0.0002 | 29.4[16.3,42.5] <0.0001 | − | |
| PP-NRS4達成e) | 12週時点のPP-NRS4達成率a) | 52.6(40/76) | 55.4(41/74) | 29.8(25/84) |
| プラセボ群との差[95%信頼区間]b) | 22.8[8.0,37.7] | 25.6[10.6,40.6] | − |
副作用発現頻度は、本剤200mg投与群で33.0%(31/94例)及び本剤100mg投与群では20.0%(19/95例)であった。主な副作用は本剤200mg投与群では悪心16.0%(15/94例)、浮動性めまい6.4%(6/94例)及び頭痛6.4%(6/94例)、本剤100mg投与群では悪心5.3%(5/95例)及び毛包炎3.2%(3/95例)であった27)。
18. 薬効薬理
18.1 作用機序
アブロシチニブはATPとの結合を遮断することにより、JAKを選択的かつ可逆的に阻害する経口投与が可能な低分子である。
18.2 JAK阻害活性
単離酵素を用いて4種類のJAKアイソフォームに対するアブロシチニブの阻害能を測定したところ、JAK1、JAK2、JAK3及びTYK2に対するIC50値はそれぞれ29.2nmol/L、803nmol/L、10,000nmol/L超及び1250nmol/Lであった28)(in vitro)。
JAKアイソフォームが介在してシグナル伝達が行われる細胞内では、JAK1が介在する種々のSTATのリン酸化を阻害(IC50値:32.5〜1690nmol/L)し、JAK2のみが介在するSTAT5のリン酸化を阻害(IC50値:794〜7780nmol/L)した29)(in vitro)。未変化体と2つの活性代謝物のサイトカインシグナル伝達の阻害は同等であった30)。[16.4参照]
JAKアイソフォームが介在してシグナル伝達が行われる細胞内では、JAK1が介在する種々のSTATのリン酸化を阻害(IC50値:32.5〜1690nmol/L)し、JAK2のみが介在するSTAT5のリン酸化を阻害(IC50値:794〜7780nmol/L)した29)(in vitro)。未変化体と2つの活性代謝物のサイトカインシグナル伝達の阻害は同等であった30)。[16.4参照]
19. 有効成分に関する理化学的知見
19.1. アブロシチニブ
| 一般的名称 | アブロシチニブ |
|---|---|
| 一般的名称(欧名) | Abrocitinib |
| 化学名 | N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfonamide |
| 分子式 | C14H21N5O2S |
| 分子量 | 323.41 |
| 物理化学的性状 | 白色〜微紫紅色の粉末である。N,N-ジメチルアセトアミドに溶けやすく、メタノール又はエタノール(99.5)に溶けにくく、水にほとんど溶けない。 |
| 分配係数 | (logP):1.66(1-オクタノール/水) |
| KEGG DRUG |
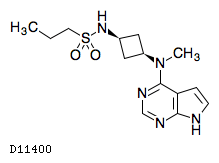
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
<サイバインコ錠50mg>
14錠[7錠(PTP)×2]
<サイバインコ錠100mg>
14錠[7錠(PTP)×2]
<サイバインコ錠200mg>
14錠[7錠(PTP)×2]
23. 主要文献
- 生殖発生毒性試験(2021年9月27日承認CTD2.4.4.8、2.6.6.7)
- 乳汁排泄試験(2021年9月27日承認CTD2.6.4.6.3)
- ラットがん原性試験(2021年9月27日承認CTD2.4.4.8、2.6.6.6.2.1)
- 反復投与毒性試験(2021年9月27日承認CTD2.6.6.4、2.6.6.10)
- フルコナゾール及びフルボキサミンとの薬物相互作用(2021年9月27日承認CTD2.7.2.2.2.3.1)
- プロベネシドとの薬物相互作用(2021年9月27日承認CTD2.7.2.2.2.3.3)
- 日本人と外国人AD患者の薬物動態の比較(2021年9月27日承認CTD2.7.2.3.3.4.2)
- マスバランス及び絶対的バイオアベイラビリティ(2021年9月27日承認CTD2.7.2.2.2.2.1)
- 食事の影響(2021年9月27日承認CTD2.7.2.2.2.2.3)
- ヒト血漿タンパク結合率(In vitro)(2021年9月27日承認CTD2.7.2.2.1.2)
- ヒトにおける代謝(In vitro)(2021年9月27日承認CTD2.7.2.2.1.3)
- 排泄(2021年9月27日承認CTD2.7.2.3.1.4)
- 腎機能障害の影響(2021年9月27日承認CTD2.7.2.3.3.2)
- 肝機能障害の影響(2021年9月27日承認CTD2.7.2.2.2.5.1)
- リファンピシンとの薬物相互作用(2021年9月27日承認CTD2.7.2.2.2.3.2)
- CYP阻害作用(In vitro)(2021年9月27日承認CTD2.7.2.2.1.4.1)
- CYP誘導作用(In vitro)(2021年9月27日承認CTD2.7.2.2.1.4.3)
- トランスポータを介した相互作用(In vitro)(2021年9月27日承認CTD2.7.2.2.1.4.6)
- 経口避妊薬との薬物相互作用(2021年9月27日承認CTD2.7.2.2.2.4.1)
- ミダゾラムとの薬物相互作用(2021年9月27日承認CTD2.7.2.2.2.4.2)
- ダビガトランとの薬物相互作用(2021年9月27日承認CTD2.7.2.2.2.4.3)
- ロスバスタチンとの薬物相互作用(2021年9月27日承認CTD2.7.2.2.2.4.4)
- メトホルミンとの薬物相互作用(2021年9月27日承認CTD2.7.2.2.2.4.5)
- 社内資料:カフェイン、エファビレンツ及びオメプラゾールとの薬物相互作用
- 国際共同第III相試験単剤投与試験(B7451013試験)(2021年9月27日承認CTD2.7.6)
- 国際共同第III相試験外用剤併用投与試験(成人)(B7451029試験)(2021年9月27日承認CTD2.7.6)
- 国際共同第III相試験外用剤併用投与試験(青少年)(B7451036試験)(2021年9月27日承認CTD2.7.6)
- 効力を裏付ける試験(2021年9月27日承認CTD2.6.2.2.1.1.1)
- ヒト全血及び各種細胞におけるサイトカイン誘発性STATリン酸化に対する阻害活性(2021年9月27日承認CTD2.6.2.2.1.2、2.6.2.2.1.3、2.6.2.2.1.4、2.6.2.2.1.5、2.6.2.2.1.6)
- アブロシチニブ代謝物の細胞レベルでの阻害活性(2021年9月27日承認CTD2.6.2.2.1.10)
24. 文献請求先及び問い合わせ先
文献請求先
ファイザー株式会社
製品情報センター
〒151-8589
東京都渋谷区代々木3-22-7
電話:学術情報ダイヤル 0120-664-467
FAX:03-3379-3053
製品情報問い合わせ先
ファイザー株式会社
製品情報センター
〒151-8589
東京都渋谷区代々木3-22-7
電話:学術情報ダイヤル 0120-664-467
FAX:03-3379-3053
26. 製造販売業者等
26.1 製造販売元
ファイザー株式会社
東京都渋谷区代々木3-22-7