医薬品情報
| 総称名 | ルプキネス |
|---|---|
| 一般名 | ボクロスポリン |
| 欧文一般名 | voclosporin |
| 製剤名 | ボクロスポリンカプセル |
| 薬効分類名 | 免疫抑制剤 カルシニューリンインヒビター |
| 薬効分類番号 | 3999 |
| ATCコード | L04AD03 |
| KEGG DRUG |
D09033
ボクロスポリン
|
| KEGG DGROUP |
DG03129
カルシニュリン阻害薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年2月 改訂(第4版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ルプキネスカプセル7.9mg | Lupkynis Capsules 7.9mg | 大塚製薬 | 3999065M1025 | 778.6円/カプセル | 劇薬, 処方箋医薬品注) |
1. 警告
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
5. 効能または効果に関連する注意
5.1 本剤投与により腎機能が悪化するおそれがあることから、eGFRが45mL/min/1.73m2以下の患者では、投与の必要性を慎重に判断し、eGFRが30mL/min/1.73m2未満の患者では可能な限り投与を避けること。eGFRが45mL/min/1.73m2以下の患者を対象とした有効性及び安全性を指標とした臨床試験は実施していない。[7.2、7.5、8.2、9.2.1、9.2.2参照]
5.2 「17.臨床成績」の項の内容を熟知し、本剤の臨床試験の投与対象、有効性及び安全性を十分に理解した上で、診療ガイドライン等の最新の情報を参考に、本剤の投与が適切と判断される患者に投与すること。[17.1.1参照]
6. 用法及び用量
通常、成人にはボクロスポリンとして1回23.7mgを1日2回経口投与する。なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
7.1 本剤の投与開始時は、原則として、副腎皮質ステロイド剤及びミコフェノール酸 モフェチルを併用すること。[17.1.1参照]
7.2 重度の腎機能障害患者(eGFR 30mL/min/1.73m2未満)への投与は可能な限り避け、やむを得ず投与する場合は、1回15.8mgを1日2回投与すること。[5.1、8.2、9.2.1、11.1.2、16.6.1参照]
7.5 腎機能が悪化した場合、以下を目安に、本剤を減量又は中止すること。[5.1、8.2、9.2.1、9.2.2、11.1.2、16.6.1参照]
・eGFRが60mL/min/1.73m2未満で、投与開始時から20%超低下した場合、1回7.9mg(1日量として15.8mg)を減量すること。減量後は、2週間以内にeGFR値を確認し、20%超の低下が持続する場合は、さらに1回7.9mg(1日量として15.8mg)を減量すること。
・eGFRが60mL/min/1.73m2未満で、投与開始時から30%超低下した場合、本剤の投与を中止すること。
7.6 血圧が上昇し、降圧剤等による適切な治療を行っても十分にコントロールできない場合は、本剤の投与を中止すること。[8.3参照]
7.7 投与開始後6箇月以内に治療の効果を確認し、投与継続の要否を検討すること。
8. 重要な基本的注意
8.3 血圧が上昇することがあるため、定期的に血圧を確認し、血圧が上昇した場合は降圧剤等による適切な治療を行うこと。[7.6参照]
8.4 痙攣発作、振戦、可逆性後白質脳症症候群(PRES)等の神経症状があらわれるおそれがあるため、本剤投与中は定期的に患者の状態を観察し、症状が認められた場合には本剤を減量又は中止すること。
8.5 本剤を含むカルシニューリン阻害薬による、重篤な高カリウム血症が報告されているため、本剤投与中は血清カリウム濃度を定期的に測定すること。[10.2参照]
8.6 カルシニューリン阻害薬による、高血糖が報告されているため、本剤投与中は定期的に血糖値等を確認すること。
8.7 過度の免疫抑制により、リンパ腫及び他の悪性腫瘍が発現するおそれがあるので、十分注意すること。[15.1.1参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 QT延長のおそれ又はその既往歴のある患者
低カリウム血症等のQT延長のリスク因子を有する患者においてQT延長が起こるおそれがある。[10.2参照]
9.2 腎機能障害患者
9.2.1 重度の腎機能障害(eGFR 30mL/min/1.73m2未満)のある患者
9.2.2 中等度の腎機能障害(eGFR 30mL/min/1.73m2以上45mL/min/1.73m2以下)のある患者
9.3 肝機能障害患者
9.3.1 重度の肝機能障害のある患者(Child-Pugh分類C)
可能な限り投与を避けること。血中濃度が上昇するおそれがある。重度の肝機能障害のある患者を対象とした臨床試験は実施していない。
9.3.2 軽度及び中等度の肝機能障害のある患者(Child-Pugh分類A及びB)
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。動物実験では、妊娠ラットに経口投与した場合、体表面積換算で臨床用量注)の約5倍(4mg/kg/日)で胎児体重の低値及び胎児の骨化遅延が認められた。また、妊娠ウサギに経口投与した場合、体表面積換算で臨床用量注)の約2倍(1.6mg/kg/日)で胎児体重の低値が、約8.2倍(6.5mg/kg/日)で胎児の胸骨未骨化等が認められた1)。
注)本剤7.9mgを60kgの患者に1回23.7mgを1日2回経口投与したときの投与量(0.79mg/kg/日)
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。ヒトで乳汁中への移行が報告されている2)。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
患者の状態を観察しながら慎重に投与すること。一般に生理機能が低下している。
10. 相互作用
相互作用序文
薬物代謝酵素用語
CYP3A4
薬物代謝酵素用語
P糖蛋白
薬物代謝酵素用語
有機アニオン輸送ポリペプチド(OATP)1B1
薬物代謝酵素用語
OATP1B3
10.1 併用禁忌
| 強いCYP3A4阻害作用を有する薬剤 アゾール系抗真菌剤 イトラコナゾール(イトリゾール) ボリコナゾール(ブイフェンド) ポサコナゾール(ノクサフィル) リトナビル含有製剤(ノービア、パキロビッド、カレトラ) アタザナビル硫酸塩(レイアタッツ) ダルナビル エタノール付加物(プリジスタ、プリジスタナイーブ) ホスアンプレナビルカルシウム水和物(レクシヴァ) コビシスタット含有製剤(ゲンボイヤ、プレジコビックス、シムツーザ) クラリスロマイシン含有製剤(クラリシッド、クラリス、ボノサップ、ラベキュア) セリチニブ(ジカディア) エンシトレルビル フマル酸(ゾコーバ) [2.2、16.7.1参照] | 代謝酵素の阻害により、本剤の作用が増強するおそれがある。 | 本剤の代謝酵素であるCYP3A4を阻害し、本剤の血中濃度を上昇させる。 |
| 生ワクチン 乾燥弱毒生麻しんワクチン 乾燥弱毒生風しんワクチン 乾燥BCG等 [2.3参照] | 類薬による免疫抑制下で、生ワクチン接種により発症したとの報告がある。 | 免疫抑制作用により発症の可能性が増加する。 |
10.2 併用注意
| 中程度のCYP3A4阻害作用を有する薬剤 フルコナゾール、ジルチアゼム、シメチジン、ベラパミル等 [7.4、16.7.3参照] | 代謝酵素の阻害により、本剤の作用が増強するおそれがあるので、本剤を減量すること。 | 本剤の代謝酵素であるCYP3A4を阻害し、本剤の血中濃度を上昇させる。 |
| グレープフルーツ含有食品 [7.4、16.7.3参照] | 代謝酵素の阻害により、本剤の作用が増強するおそれがあるので、本剤を減量すること。 | 本剤の代謝酵素であるCYP3A4を阻害し、本剤の血中濃度を上昇させる。 |
| 中程度以上のCYP3A4誘導作用を有する薬剤 リファンピシン、エファビレンツ等 セイヨウオトギリソウ(St.John's Wort、セントジョーンズワート)含有食品 [16.7.2参照] | 代謝酵素の誘導により、本剤の作用が減弱するおそれがある。 | 本剤の代謝酵素であるCYP3A4を誘導し、本剤の血中濃度を低下させる。 |
| HMG-CoA還元酵素阻害剤 シンバスタチン等 [16.7.5参照] | 本剤によりHMG-CoA還元酵素阻害剤の血中濃度が上昇し、副作用の発現頻度が増加するおそれがある。 | 本剤はOATP1B1/3を阻害しHMG-CoA還元酵素阻害剤の血中濃度を上昇させるおそれがある。 |
| ジゴキシン [16.7.4参照] | 本剤によりジゴキシンの作用が増強されるおそれがあるので、ジゴキシンを減量するなど慎重に投与すること。 | 本剤はP糖蛋白を阻害し、ジゴキシンの血漿中濃度を上昇させる。 |
| カリウム製剤 カリウム保持性利尿剤 スピロノラクトン、トリアムテレン等 抗アルドステロン薬 エプレレノン等 アンジオテンシン変換酵素阻害薬 エナラプリルマレイン酸塩等 アンジオテンシンII受容体拮抗薬 ロサルタンカリウム等 レニン阻害薬 アリスキレンフマル酸塩等 非ステロイド性消炎鎮痛剤 ジクロフェナク ナプロキセン スリンダク インドメタシン等 ジゴキシン β-遮断剤 ヘパリン [8.5参照] | 高カリウム血症があらわれるおそれがあるので、血清カリウム値に注意すること。 | 高カリウム血症の副作用が相互に増強される。 |
| 腎毒性のある薬剤 アムホテリシンB、アミノ糖系抗生物質、スルファメトキサゾール・トリメトプリム、非ステロイド性抗炎症剤等 | 腎障害が発現することがある。 | 腎毒性が相互に増強される。 |
| 不活化ワクチン インフルエンザワクチン等 | 不活化ワクチンの作用を減弱させることがある。 | 免疫抑制作用によりワクチンに対する免疫が得られないおそれがある。 |
| PUVA療法を含む紫外線療法 [15.1.1参照] | PUVA療法を含む紫外線療法との併用は皮膚癌発現のリスクを高める危険性があるため、やむを得ず併用する場合は定期的に皮膚癌又は前癌病変の有無を観察すること。 | PUVA療法により皮膚癌が発生したとの報告があり、本剤併用による免疫抑制下では皮膚癌の発現を促進する可能性がある。 |
| 免疫抑制作用を有する薬剤 免疫抑制剤 副腎皮質ホルモン剤等 抗リウマチ薬(DMARD) メトトレキサート等 [15.1.1参照] | 過度の免疫抑制が起こることがある。 | ともに免疫抑制作用を有する。 |
| QT延長を起こすことが知られている薬剤 ヒドロキシクロロキン アジスロマイシン シプロフロキサシン等 [9.1.1参照] | QT延長を起こすおそれがある。 | これらの薬剤では単独投与でもQT延長がみられている。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 重篤な感染症(10.1%)
発現頻度は海外第II相及び国際共同第III相試験に基づく。
11.2 その他の副作用
| 10%以上 | 10%未満 | 頻度不明 | |
| 感染症 | 上気道感染(24.0%) | インフルエンザ、帯状疱疹 | |
| 血液 | 貧血 | ||
| 免疫系 | 過敏症 | ||
| 代謝 | 高カリウム血症、食欲減退 | ||
| 精神神経系 | 頭痛 | 痙攣発作、振戦 | |
| 循環器 | 高血圧(20.6%) | ||
| 呼吸器 | 咳嗽 | ||
| 消化器 | 下痢、腹痛 | 悪心、歯肉増殖、消化不良 | |
| 皮膚 | 脱毛症、多毛症 | ||
| 腎および尿路 | 糸球体濾過率減少(26.2%) |
13. 過量投与
13.1 症状
本剤の過量投与の報告は限られているが、過量投与により、振戦、頻脈が現れたとの報告がある。
13.2 処置
特異的な解毒剤はない。
14. 適用上の注意
14.1 薬剤交付時の注意
14.1.1 PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
14.1.2 服用直前にPTPシートからカプセルを取り出すよう指導すること。[20.参照]
14.1.3 本剤はカプセルを開けたり、つぶしたり、分割せずそのまま水で服用するよう指導すること。
15. その他の注意
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人にボクロスポリン23.7mgを絶食時及び食後に単回経口投与した時の血中濃度推移及び薬物動態パラメータを図16-1及び表16-1に示す4)。
図16-1 健康成人におけるボクロスポリン単回経口投与時の血中濃度推移
| 食事条件 | 例数 | tmax(h) | Cmax(ng/mL) | AUC∞(ng・h/mL) | t1/2(h) |
| 絶食時 | 16 | 1.50(1.00〜2.50) | 115±19.9 | 455±110a | 13.1±4.6a |
| 食後 | 16 | 2.00(1.50〜4.00) | 110±33.4 | 522±179 | 14.2±6.8 |
16.1.2 反復投与
健康成人にボクロスポリン0.25mg/kg注)(平均体重を基に換算すると17.1mg)、0.5mg/kg注)(平均体重を基に換算すると35.2mg)、1.0mg/kg注)(平均体重を基に換算すると70.6mg)又は1.5mg/kg注)(平均体重を基に換算すると95.9mg)を1日目の朝に空腹時単回経口投与後、3日目から12日目までの10日間1日2回(朝夜、約12時間ごと)空腹時反復経口投与した後13日目の朝に空腹時単回経口投与した時、ボクロスポリンの血中濃度は6日間投与後に定常状態に達した。薬物動態パラメータを表16-2に示す5)。
| 投与量 | 例数 | tmax(h) | Cmax(ng/mL) | AUC24hb(ng・h/mL) | t1/2(h) |
| 17.1mg (0.25mg/kg) | 8 | 1.57(1.08〜2.13) | 70.3±19.6 | 372±104 | 27.1±4.27 |
| 35.2mg (0.5mg/kg) | 8 | 1.56(1.08〜4.07) | 160±56.7 | 921±399 | 29.6±4.58a |
| 70.6mg (1.0mg/kg) | 8 | 2.12(1.57〜2.55) | 416±46.7 | 2,510±317 | 29.9±4.00 |
| 95.9mg (1.5mg/kg) | 8 | 2.00(1.50〜3.00) | 619±82.5 | 4,390±945 | 30.6±3.81a |
16.2 吸収
16.2.1 食事の影響
健康成人にボクロスポリン23.7mgを単回経口投与した時、絶食時投与に比べ食後(高脂肪食)投与ではCmax及びAUCはそれぞれ0.91倍及び1.14倍であった4)。
16.3 分布
ヒト血漿蛋白結合率は、96.97%であった6)(in vitro、平衡透析法)。
16.4 代謝
16.5 排泄
健康成人に14C標識ボクロスポリン70mg注)を単回経口投与した時、糞中及び尿中にそれぞれ投与した放射能の92.7%及び2.1%が排泄された。未変化体の糞中及び尿中への排泄率はそれぞれ5%及び0.25%であった8)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
16.6.2 肝機能障害患者
16.6.3 性別
ループス腎炎患者においてボクロスポリンの薬物動態に性別による臨床的に意味のある影響は認められなかった11)(外国人データを含む母集団解析)。
16.7 薬物相互作用
16.7.1 ケトコナゾール
16.7.2 リファンピシン
16.7.3 ベラパミル
16.7.4 ジゴキシン
16.7.5 シンバスタチン
16.7.6 その他
・全身性エリテマトーデス患者において、ミコフェノール酸モフェチル(MMF)1gとボクロスポリン23.7mg1日2回の併用投与は、ミコフェノール酸(MPA)血中濃度に臨床的に有意な影響を及ぼさなかった17)(外国人データ)。
・健康成人において、ボクロスポリン0.4mg/kg注)1日2回とCYP3A4基質であるミダゾラム7.5mgの併用投与は、ミダゾラムのCmax及びAUCに顕著な影響を与えず、それぞれ単独投与時と比較して約0.89倍及び1.02倍であった18)(外国人データ)。
注)本剤の承認された用法及び用量は、通常1回23.7mgを1日2回である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第III相試験
eGFRが45mL/min/1.73m2超の活動性ループス腎炎患者[ISN/RPS分類ClassIII、IV(単独又はClassVとの複合)でUPCRが1.5mg/mg以上、又はClassVでUPCRが2.0mg/mg以上]357例(日本人患者13例を含む)を対象としたプラセボ対照二重盲検比較試験において、ミコフェノール酸 モフェチル及びステロイドの併用下で、本剤23.7mg又はプラセボを1日2回52週間経口投与した。主要評価項目である52週時点の腎奏効率注1)は、本剤群40.8%、プラセボ群22.5%であり、プラセボ群と比較して統計学的に有意に高かった(p<0.001)(表17-1)21)。
| プラセボ群 (178例) | 本剤群 (179例) | プラセボ群とのオッズ比 [95%信頼区間] p値 | |
| 52週時点の腎奏効率 | 22.5% | 40.8% | 2.65 [1.64,4.27] p<0.001 |
17.1.2 国際共同長期投与試験
国際共同第III相試験で52週間(12箇月)投与を完了したループス腎炎患者216例(日本人患者6例を含む)を対象としたプラセボ対照二重盲検長期投与試験において、ミコフェノール酸 モフェチル及びステロイドの併用下で、本剤23.7mg又はプラセボを1日2回24箇月間、計36箇月間経口投与した時、投与開始12箇月以降6箇月ごとの腎奏効率注1)の推移は表17-2のとおりであった22)。
| プラセボ群 (100例) | 本剤群 (116例) | プラセボ群とのオッズ比 [95%信頼区間] | |
| 12箇月時点の腎奏効率 | 34.0% | 52.6% | 2.30 [1.30,4.05] |
| 18箇月時点の腎奏効率 | 46.0% | 63.8% | 2.19 [1.25,3.83] |
| 24箇月時点の腎奏効率 | 43.0% | 56.0% | 1.81 [1.04,3.16] |
| 30箇月時点の腎奏効率 | 42.0% | 59.5% | 2.24 [1.28,3.92] |
| 36箇月時点の腎奏効率 | 39.0% | 50.9% | 1.74 [1.00,3.03] |
副作用発現頻度は、本剤群で116例中28例(24.1%)であった。主な副作用は、糸球体濾過率減少8例(6.9%)であった。
注1)[1]〜[5]全てを満たすことを基に判定委員会で判定。[1]UPCRが0.5mg/mg以下である、[2]eGFRが60mL/min/1.73m2以上である、又はeGFRのベースラインから20%を超える低下が確認されない、[3]ループス腎炎治療のため、救済薬投与を受けていない、[4]腎奏効評価前8週間に投与されたプレドニゾン量が3日間以上連続又は合計7日間以上で10mgを超えない、[5]52週時点で試験を中止していない。
17.3 その他
17.3.1 QT間隔に対する影響(ISA03-11試験)
外国人健康成人を対象に、本剤0.5、1.5、3.0、4.5mg/kg注2)を単回経口投与したときのQTc間隔の変化量のプラセボとの差の最大値(95%片側信頼区間の上限値)は、それぞれ6.4(11.6)ms、14.9(20.1)ms、25.7(30.9)ms及び34.6(39.8)msであり、いずれの用量においてもQTc間隔の延長が認められた。また、モキシフロキサシン400mgを単回経口投与したときにQTc間隔の延長が認められた23)。
17.3.2 QT間隔に対する影響(ISA05-03試験)
外国人健康成人を対象に、本剤0.3、0.5、1.5mg/kg注2)を7日間反復経口投与したときのQTc間隔の変化量のプラセボとの差の最大値(95%片側信頼区間の上限値)は、それぞれ0.8(4.7)ms、2.4(6.2)ms及び2.8(6.9)msであり、いずれの用量においてもQTc間隔が延長する傾向は認められなかった。一方、モキシフロキサシン400mgを単回経口投与したときにQTc間隔の延長が認められた23)。
注2)本剤の承認された用法及び用量は、通常1回23.7mgを1日2回である。
18. 薬効薬理
18.1 作用機序
ボクロスポリンは、T細胞においてシクロフィリンと複合体を形成し、カルシニューリンに結合することでカルシニューリンを阻害する。これによりリンパ球増殖、T細胞サイトカイン産生、及びT細胞活性化表面抗原の発現が抑制され、免疫抑制作用を示す。
18.2 カルシニューリン阻害作用
ボクロスポリンはカルシニューリン活性の阻害作用を示した24)(in vitro)。
18.3 リンパ球増殖抑制作用
ボクロスポリンは、リンパ球増殖抑制作用を示した25)(in vitro)。
18.4 心移植モデルにおける免疫抑制作用
ボクロスポリンは、心移植モデルラットにおいて異種移植した心臓の移植片生着期間の延長効果を示した26)。
19. 有効成分に関する理化学的知見
19.1. ボクロスポリン
| 一般的名称 | ボクロスポリン |
|---|---|
| 一般的名称(欧名) | voclosporin |
| 化学名 | Cyclo{[(2S,3R,4R,6E)-3-hydroxy-4-methyl-2-(methylamino)nona-6,8-dienoyl]-L-2-aminobutanoyl-N-methylglycyl-N-methyl-L-leucyl-L-valyl-N-methyl-L-leucyl-L-alanyl-D-alanyl-N-methyl-L-leucyl-N-methyl-L-leucyl-N-methyl-L-valyl} |
| 分子式 | C63H111N11O12 |
| 分子量 | 1,214.62 |
| 物理化学的性状 | 白色〜オフホワイトの粉末又は塊を含む粉末である。アセトニトリル、メタノール、エタノール(99.5)又はアセトンに溶けやすく、水又はヘプタンにほとんど溶けない。 |
| 分配係数 | log P=6.23(1-オクタノール/水、低速攪拌法) |
| KEGG DRUG |
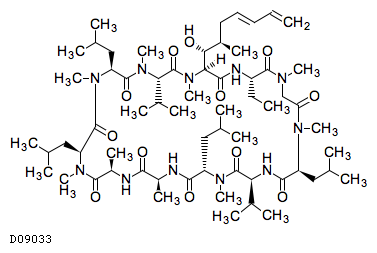
|
20. 取扱い上の注意
吸湿性を有するためPTP包装のまま保存すること。[14.1.2参照]
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
PTP
100カプセル(10カプセル×10)
23. 主要文献
- 社内資料:毒性試験(2024年9月24日承認、CTD2.6.6)
- 社内資料:ヒト乳汁中移行に関する試験(2024年9月24日承認、CTD2.7.6.2)
- Penn,I., Drug Saf., 23 (2), 101-113, (2000) »PubMed
- 社内資料:健康成人における食事の影響試験(2024年9月24日承認、CTD2.7.6.1)
- 社内資料:健康成人における薬物動態試験(2024年9月24日承認、CTD2.7.6.2)
- 社内資料:In vitro血漿蛋白結合率試験(2024年9月24日承認、CTD2.6.4.4)
- 社内資料:In vitro代謝酵素同定試験(2024年9月24日承認、CTD2.6.4.5)
- 社内資料:14C標識ボクロスポリンを用いた薬物動態試験(2024年9月24日承認、CTD2.7.6.2)
- 社内資料:腎機能障害患者を対象とした薬物動態試験(2024年9月24日承認、CTD2.7.6.2)
- 社内資料:肝機能障害患者を対象とした薬物動態試験(2024年9月24日承認、CTD2.7.6.2)
- 社内資料:ループス腎炎患者を対象とした母集団薬物動態解析(2024年9月24日承認、CTD2.7.2.3)
- 社内資料:経口ケトコナゾールとの相互作用試験(2024年9月24日承認、CTD2.7.6.2)
- 社内資料:リファンピシン(反復)との相互作用試験(2024年9月24日承認、CTD2.7.6.2)
- 社内資料:ベラパミルとの相互作用試験(2024年9月24日承認、CTD2.7.6.2)
- 社内資料:ジゴキシンとの相互作用試験(2024年9月24日承認、CTD2.7.6.2)
- 社内資料:シンバスタチンとの相互作用試験(2024年9月24日承認、CTD2.7.6.2)
- 社内資料:ミコフェノール酸 モフェチルとの相互作用試験(2024年9月24日承認、CTD2.7.6.2)
- 社内資料:ミダゾラムとの相互作用試験(2024年9月24日承認、CTD2.7.6.2)
- 社内資料:In vitroトランスポーター基質性試験(2024年9月24日承認、CTD2.6.4.7)
- 社内資料:In vitroトランスポーター阻害試験(2024年9月24日承認、CTD2.6.4.7)
- 社内資料:ループス腎炎患者を対象とした第III相試験(2024年9月24日承認、CTD2.7.6.4)
- 社内資料:ループス腎炎患者を対象とした第III相長期試験(2024年9月24日承認、CTD2.7.6.4)
- 社内資料:健康成人における単回及び反復投与時のQTcF評価(2024年9月24日承認、CTD2.7.2.2)
- 社内資料:In vitroヒトカルシニューリン阻害アッセイ(2024年9月24日承認、CTD2.6.2.2)
- 社内資料:In vitroヒトリンパ球増殖アッセイ(2024年9月24日承認、CTD2.6.2.2)
- 社内資料:ラット異所性心移植モデルにおける有効性(2024年9月24日承認、CTD2.6.2.2)
24. 文献請求先及び問い合わせ先
文献請求先
大塚製薬株式会社
医薬情報センター
〒108-8242
東京都港区港南2-16-4 品川グランドセントラルタワー
電話:0120-189-840
FAX:03-6717-1414
製品情報問い合わせ先
大塚製薬株式会社
医薬情報センター
〒108-8242
東京都港区港南2-16-4 品川グランドセントラルタワー
電話:0120-189-840
FAX:03-6717-1414
26. 製造販売業者等
26.1 製造販売元
大塚製薬株式会社
東京都千代田区神田司町2-9